
The lecture below can be accessed on the Disease Management section of the Cleveland Clinic, under Allergy and Immunology (to go to this link and see others in the series, please click here)
Dilated and Restrictive Cardiomyopathies
Corinne Bott-Silverman, MD
Baran Aksut, MD
Published: November 2015
Cardiomyopathies are diseases of the myocardium associated with cardiac dysfunction.1 Table 1 lists the five types of cardiomyopathy: dilated, hypertrophicn restrictive arrhythmogenic right ventricular, and unclassified. Many conditions manifest as one type of cardiomyopathy and progress to another. For example, hypertensive heart disease can begin with a hypertrophic pattern and subsequently become a dilated cardiomyopathy. In addition, some conditions have features of more than one type of cardiomyopathy. For example, sarcoidosis can have features of restrictive and dilated cardiomyopathy at different times in the course of the disease.
Cardiomyopathy often results in the heart failure syndrome, with a number of systemic manifestations. On the other hand, many systemic conditions have cardiac involvement and manifest primarily as heart failure. The cardiomyopathies represent a diverse group of conditions whose final common pathway is myocardial dysfunction. With few exceptions, histologic findings are nonspecific and include myocyte hypertrophy, cellular necrosis, and fibrosis.
There are many known causes of cardiomyopathy. Many systemic diseases have myocardial involvement, which can range from mild to severe (Table 2). The most common cause of cardiomyopathy in developed countries is ischemic cardiomyopathy. In other geographic regions, such as equatorial Africa, infiltrative disease is the leading cause of cardiomyopathy.
In this chapter, discussion is confined to the definition, prevalence, signs and symptoms, and diagnosis of cardiomyopathies, with the exclusion of hypertrophic cardiomyopathy, which is discussed separately.
Dilated Cardiomyopathy
Definition
Dilated cardiomyopathy may be defined as an ejection fraction of less than 40% in the presence of increased left ventricular dimension (left ventricular end-diastolic size >115% of that calculated for age and body surface area). Increased left ventricular dimensions in the presence of preserved systolic function may be a precursor to the development of systolic dysfunction in certain patients. Pharmacologic intervention with angiotensin-converting enzyme inhibitors/angiotensin receptor blockers, beta blockers, and aldosterone antagonists might prevent progression to heart failure in some of these patients.
Prevalence
It is difficult to assess the prevalence of cardiomyopathy accurately. Many cases go undiagnosed and patients with undiagnosed cardiomyopathy can present with sudden cardiac death. Strict diagnostic criteria are lacking. Approximately 5 million Americans have symptomatic heart failure, but it has been estimated that 50 million Americans fulfill American Heart Association–American College of Cardiology definitions of classes A and B heart failure (Table 3)2 and are either at risk for or have established structural heart disease in the absence of heart failure symptoms. It is unclear how many people fall into stages B, C, and D combined (those with structural heart disease, with or without heart failure symptoms); most of these people have cardiomyopathies. The estimated prevalence of idiopathic dilated cardiomyopathy is 0.4 per 1,000 of the general population. However, in the future, as more causes are elucidated and more patients are found to have genetic or familial cardiomyopathy, the number of patients with idiopathic disease, a diagnosis of exclusion, will decrease.
Pathophysiology
Dilated cardiomyopathy represents the final common morphologic outcome of various biologic insults. The combination of myocyte injury and necrosis associated with myocardial fibrosis results in impaired mechanical function. Many cases are a result of direct toxicity (eg, alcohol) or mechanical insults (eg, chronic volume overload in mitral valvular regurgitation). With myocyte failure and cytoskeletal uncoupling, the chambers become dilated. According to Laplace’s law, increased diameter increases wall stress and causes further mechanical disadvantage. Thus, myocardial dysfunction can cause a vicious cycle leading to more myocardial dysfunction in a process termed adverse ventricular remodeling, which is an important therapeutic target.3
No specific genetic abnormality is known to cause dilated cardiomyopathy though numerous genetic abnormalities have been found. There are many putative mechanisms in the development of familial cardiomyopathy beyond the scope of this chapter. All forms of Mendelian inheritance have been observed, including autosomal dominant, recessive, X-linked, and mitochondrial (matrilinear).4
Table 1: World Health Organization Classification of Cardiomyopathies
| Type of Cardiomyopathy | Features | Causative Factors |
|---|---|---|
| Dilated | Dilated left or both ventricle(s), with impaired contraction | Ischemic, idiopathic, familial-genetic, immune, alcoholic, toxic, valvular |
| Hypertrophic | Left or right ventricular hypertrophy, or both | Familial, with autosomal dominant inheritance |
| Restrictive | Restrictive filling and reduced diastolic filling of one or both ventricles; normal or near-normal systolic function | Idiopathic, amyloidosis, endomyocardial fibrosis |
| Arrhythmogenic right ventricular cardiomyopathy | Fibrofatty replacement of right ventricular myocardium, Uhl’s anomaly (parchment heart) | Unknown; familial, usually autosomal dominant inheritance, with incomplete penetrance; possible autosomal recessive inheritance; rare forms associated with typical phenotype (eg, Naxos disease) |
| Unclassified | Not typical for previous four groups | Fibroelastosis, noncompacted myocardium, systolic dysfunction with minimal dilation, mitochondrial disease |
Data from Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation 1996; 93:841–842.
Table 2: Causes of Cardiomyopathy and Associated Disorders
| Cause | Disorder |
|---|---|
| Cardiovascular | |
| Ischemic heart disease | |
| Hypertension | |
| Valvular heart disease | |
| Idiopathic dilated cardiomyopathy | |
| Idiopathic restrictive cardiomyopathy | |
| Cardiac amyloidosis | |
| Metabolic | |
| Starvation, vitamin deficiency | |
| Diabetes, hypothyroidism and hyperthyroidism, acromegaly, pheochromocytoma | |
| Glycogen storage disease | |
| Infectious, inflammatory | |
| Secondary amyloidosis, sarcoidosis | |
| Acute viral (Coxsackie B), HIV, hepatitis C | |
| Chagas disease (protozoal) | |
| Toxic | |
| Alcohol, cocaine, amphetamines, chemotherapy | |
| Genetic | |
| Familial dilated cardiomyopathy | |
| Familial cardiac amyloidosis | |
| Noncompation of the myocardium | |
| Systolic dysfunction without dilation | |
| Arrhythmogenic right ventricular cardiomyopathy | |
| Hemochromatosis | |
| Tachycardia | |
| Tachycardia-induced cardiomyopathy | |
| Pregnancy | |
| Peripartum cardiomyopathy | |
© 2003 The Cleveland Clinic Foundation.
Table 3: American Heart Association/American College of Cardiology Staging of Heart Failure
| Stage | Definition |
|---|---|
| A | Patients at risk of heart failure, with no structural heart disease |
| B | Patients with structural heart disease, without symptoms of heart failure |
| C | Patients with past or present heart failure symptoms |
| D | Patients with advanced disease (e.g., inotropic support) |
Data from Hunt SA, Abraham WT, Chin MH, et al. American College of Cardiology; American Heart Association Task Force on Practice Guidelines; American College of Chest Physicians; International Society for Heart and Lung Transplantation; Heart Rhythm Society: ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): Developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: Endorsed by the Heart Rhythm Society. Circulation 2005; 112:e154–e235.
Causes and Conditions Associated with Dilated Cardiomyopathy
Ischemic Cardiomyopathy
Ischemic cardiomyopathy (ICM) is the most commonly identified specific cause of dilated cardiomyopathy, accounting for more than 60% of patients with symptomatic heart failure and many more with asymptomatic left ventricular dysfunction. There are several mechanisms by which coronary artery disease can result in ICM.
Myocardial infarction (MI) causes localized myocyte necrosis, with resultant scar formation and loss of contractile function in the ventricular segment perfused by the culprit artery. In addition, the myocardium distal to the area of infarction develops increased wall stress, adverse remodeling, and chamber dilation, so that a cardiomyopathic process occurs in adjacent nonischemic areas.
Another mechanism for myocardial dysfunction is hibernation, in which areas of myocardium are chronically underperfused and metabolically less active. These areas remain metabolically intact but do not contribute to the mechanical activity of the heart. Identification of these areas and restoration of their perfusion through revascularization might improve the ejection fraction and long-term prognosis.
Additional features of ICM include the development of mitral valvular regurgitation, which may be caused by papillary muscle dysfunction or functional factors, such as failure of mitral valve leaflets to coapt in a dilated ventricle. This further increases the volume overload state, increasing myocardial energy demands and causing a vicious cycle of worsening systolic dysfunction.
Atrial and ventricular arrhythmias occur commonly in ICM and include atrial fibrillation, which can further compromise contractile function. The development of atrioventricular conduction delays with the necessity for permanent pacemaker insertion can also cause pacing-induced dyssynchrony when pacing is performed from the right ventricular apex alone.
Ischemic cardiomyopathy is generally ascribed to epicardial coronary atherosclerosis, but it can also occur in any vasculitic process (eg, Takayasu’s arteritis), congenital abnormalities (including aberrant coronary arteries), embolic conditions (eg, atrial fibrillation, endocarditis, thrombophilic states), cardiac allograft vasculopathy, and microvascular ischemia.
Idiopathic Dilated Cardiomyopathy
The term idiopathic dilated cardiomyopathy is applied to most patients with nonischemic cardiomyopathy. With progress in the field of gene analysis, it is likely that many patients with idiopathic cardiomyopathy will receive a specific molecular or genetic diagnosis in the future.
Myocarditis
Myocarditis may be a more common prelude to dilated cardiomyopathy than was once believed. There are four different clinicopathologic classifications of myocarditis — fulminant, acute, chronic active, and chronic persistent, with the acute form being the most common. The natural history of myocarditis, and more specifically acute myocarditis, is largely unknown. Myocarditis is most commonly caused by viral infections mainly Coxsackievirus B, adenovirus, or parvovirus, however it also includes other infectious agents (ie, bacterial, human immunodeficiency virus (HIV)-associated, Lyme disease, Trypanosoma cruzi) and non-infectious forms (ie, cardiotoxic drugs, collagen vascular diseases, systemic illnesses such as sarcoidosis, and rheumatic fever).5-8
Myocarditis should be suspected when patients present with otherwise unexplained cardiac abnormalities such as heart failure, cardiogenic shock, or arrhythmias. The age of onset is typically between 20 to 50 years old. Some patients have a history of a recent viral illness but the majority do not. Myocarditis should also be suspected when patients have a history of acute or subacute development of left ventricular dysfunction without an apparent etiology; when a patient presents with pericarditis (infectious or idiopathis) with cardiac biomarker elevation suggestive of myopericarditis; or when a patient presents with clinical signs and symptoms of an acute MI and a normal coronary angiogram.
Overall, approximately 50% of patients who receive a diagnosis of acute viral myocarditis develop dilated cardiomyopathy. Up to 75% of patients with non-ischemic dilated cardiomyopathy who have had a clinically recognized episode of myocarditis have genomic viral DNA persistence in myocardial samples. Despite this, endomyocardial biopsy (EMB) rarely shows myocarditis in patients with new-onset cardiomyopathy. Most have non-specific histologic findings by light microscopy. There is significant interobserver variability in the pathologic diagnosis of myocarditis.8 Therefore, the indications for biopsy are limited and in the acute setting are done only if it will change management as can be seen in patients with suspected giant cell myocarditis (GCM) where early consideration for transplantation would be given. Other than certain specific instances where one can consider immunosuppression or early transplantation, the main form of treatment for patients with suspected myocarditis is supportive care. Depending on the degree of ventricular dysfunction, in addition to medical support, mechanical support in the form of an intra-aortic balloon pump or other assist device can be given1 until the patient recovers from the acute illness. Once they have passed this acute phase, therapy focuses on the medical management of heart failure and on arrhythmia management including implantable cardioverter defibrillator (ICD) placement if indicated. It has been suggested that patients refrain from vigorous exercise due to a theoretical increased risk of myocardial inflammation and adverse remodeling that can occur.5
Hypertensive Heart Disease
Hypertensive heart disease can initially manifest as left ventricular hypertrophy with isolated diastolic dysfunction and preserved systolic function, as assessed by conventional echocardiographic techniques. Because remodeling occurs over time, the hypertrophy can progress to a dilated cardiomyopathy with systolic dysfunction. Atrial fibrillation is a common manifestation of hypertensive heart disease. Hypertensive heart disease is the leading identifiable cause of heart failure in older women.
Valvular Heart Disease
Hemodynamically significant valvular lesions, such as aortic stenosis, aortic regurgitation, and mitral regurgitation, produce pressure and volume overload states that can result in adverse ventricular remodeling and the development of systolic, diastolic, or combined myocardial dysfunction. In valvular disease, excess hemodynamic demands result in myocyte hypertrophy, subsequent chamber enlargement, and myocardial fibrosis. Chamber dilation then creates or exacerbates existing mitral or tricuspid valvular regurgitation, or both. With further chamber dilation, subendocardial ischemia and localized myocyte necrosis develop. In addition, concomitant coronary artery disease, especially with degenerative aortic stenosis, and atrial fibrillation, especially with mitral regurgitation, can cause further deterioration (See Aortic Valve Disease and Mitral Valve Disease).
Toxic Cardiomyopathies
Alcoholic cardiomyopathy may account for approximately 4% of all cardiomyopathies and men have a significantly worse prognosis.9 The average duration of heavy drinking (more than 90 g/day) is 15 years in most cohorts. Diastolic dysfunction usually precedes any evidence of systolic dysfunction. Left ventricular dilation is an early finding. Hypertension, atrial fibrillation (holiday heart), and coronary disease are more common in heavy drinkers. Identification of alcohol as a potential cause of cardiomyopathy is vital; abstinence can result in an improved ejection fraction in 50% of patients medically treated for heart failure and continued drinking can result in further deterioration of cardiac function. The mechanism of alcohol-induced cardiomyopathy is unclear but might involve disturbances in intracellular calcium transients, mitochondrial disruption, decreased myofibrillary proteins, and myocyte apoptosis. Histologic findings are nonspecific.
Cocaine and amphetamines (including 3,4-methylenedioxymethamphetamine, or ecstasy) can result in dilated cardiomyopathy with single and chronic use.10 The cause is multifactorial and includes direct myocyte toxicity, tachycardia-induced injury, hypertension, and MI.
Doxorubicin can cause cardiomyopathy with characteristic histopathologic features. Trastuzumab, used in the treatment of metastatic breast cancer, can cause a cardiomyopathy. Unlike anthracycline-induced toxicity, trastuzumab-induced cardiomyopathy usually responds to standard treatment or discontinuation of trastuzumab.11Brain natriuretic peptide (BNP) is proving useful in monitoring cardiac function in patients receiving cardiotoxic chemotherapy, because elevation of the BNP level occurs at an early stage in the condition. Hydroxychloroquine can cause skeletal and cardiac myopathies.
Peripartum cardiomyopathy is dilated cardiomyopathy arising in the last month of pregnancy or within 5 months postpartum.12 Of these cases, 75% occur in the first 2 months after delivery. Risk factors include age older than 30 years, multiparity, twin pregnancy, African descent, and a family history of peripartum cardiomyopathy.13 The cause of peripartum cardiomyopathy is unknown but may be related to reduced suppressor T cell activity, which occurs during pregnancy and can result in an autoimmune type of myocardial inflammation or activation of myocarditis. Recovery usually occurs within 6 months in 50% of patients. Patients should be advised not to have more children (See Pregnancy and Heart Disease).
Infective Cardiomyopathies
In addition to acute myocarditis (often presumed viral) discussed earlier, various other viral agents have been implicated in the development of cardiomyopathy, including HIV and hepatitis C. Trypanosoma cruzi (a protozoan) has infected 20 million people in South and Central America. Trypanosoma cruzi Infection causes Chagas’ disease, a dilated cardiomyopathy (either global or with characteristic apical aneurysm formation), in 20% to 30% of patients, acutely or over many years. Other parasitic infestations that can cause cardiomyopathy in the immunocompetent and immunocompromised patient include Toxoplasma gondii and Trichinella spiralis. Plasmodium falciparum infection (malaria) can cause parasitic coronary artery occlusion.
Tachycardia-Induced Cardiomyopathy
Prolonged exposure to rapid heart rates can induce myocardial dysfunction. Persistent or permanent atrial fibrillation induces electrical and structural remodeling of the atria. When rapidly conducted, it can cause adverse ventricular remodeling and a dilated cardiomyopathy. The diagnosis is one of exclusion and rate control or restoration of sinus rhythm can restore systolic function. Sometimes cause and effect can be difficult to determine.
Metabolic Conditions
Malnutrition, as well as selenium, carnitine, phosphate, calcium, and vitamin B deficiencies, can result in dilated cardiomyopathy. Endocrine causes include adrenocortical insufficiency, thyrotoxicosis, hypothyroidism, acromegaly, and pheochromocytoma.
Stress Cardiomyopathy (Takosubo Cardiomyopathy)
Stress or Takosubo cardiomyopathy is a syndrome of reversible ventricular systolic dysfunction precipitated by acute emotional or physiologic stress. Stress cardiomyopathy can mimic an acute MI but has recognizable electrocardiogram (ECG) and angiographic features. It is believed to be sympathetically mediated but the exact pathogenesis in unknown. Required diagnostic criteria are the absence of coronary thrombosis, wall motion abnormalities involving multiple coronary territories, and the rapid and complete recovery of systolic function with supportive and medical care.
Restrictive Cardiomyopathies
Definition
Restrictive cardiomyopathy is a disease of the myocardium characterized by restrictive filling and reduced diastolic volume of either or both ventricles, with normal or near-normal systolic function.1
Prevalence
Restrictive cardiomyopathies represent a very small fraction (less than 5%) of cardiomyopathies in the West,14 but they are more common in certain populations. For example, endomyocardial fibrosis is a relatively common cause of heart failure in equatorial Africa though ischemic heart disease is less common.
Pathophysiology
Restrictive cardiomyopathies result in impaired ventricular filling and primarily diastolic heart failure. They manifest with a clinical heart failure syndrome that is often indistinguishable from that caused by systolic dysfunction. Atrioventricular block and symptomatic bradycardias can be seen, often necessitating pacemaker insertion. Atrial fibrillation is poorly tolerated.
Restrictive cardiomyopathies may be classified as primary (eg, endomyocardial fibrosis, Löffler’s endocarditis, idiopathic restrictive cardiomyopathy) or secondary. Causes of secondary restrictive cardiomyopathy include infiltrative diseases (eg, amyloidosis, sarcoidosis, radiation carditis) and storage diseases (eg, hemochromatosis, glycogen storage disorders, Fabry’s disease). Fabry’s disease, although rare, has assumed a new importance as effective treatment has become available.15
Amyloid heart disease is classified as primary, secondary, familial, or senile. Primary amyloid heart disease is caused by overproduction of light chain immunoglobulin from a monoclonal population of plasma cells, usually associated with multiple myeloma. Secondary amyloid heart disease is associated with chronic inflammatory conditions such as Crohn’s disease, rheumatoid arthritis, tuberculosis, and Familial Mediterranean fever. Familial and senile amyloid heart disease are related to the overproduction of transthyretin. Myocardial amyloid heart disease is confirmed by EMB. The presence of near-normal left ventricular dimensions combined with increased myocardial wall thickness, particularly biventricular thickening, should arouse suspicion of an infiltrative cardiomyopathy, especially if accompanied by low-voltage QRS complexes on the ECG. Unfortunately, there is no proven treatment for amyloid heart disease and the prognosis is poor.16 Primary amyloid sometimes responds to chemotherapy directed at the associated multiple myeloma.
Cause and Conditions Associated with Restrictive Cardiomyopathies
Hemochromatosis
Hemochromatosis (bronze diabetes) is a disease that results in iron overload and deposition of iron in the sarcoplasmic reticulum of many organs, including the heart. Hemochromatosis generally follows an autosomal recessive pattern of Mendelian inheritance. The use of serum ferritin levels as a screen for this condition is reasonable. Cardiac magnetic resonance imaging (MRI) can be useful for the diagnosis of cardiac involvement. Hemochromatosis can result in a restrictive or dilated cardiomyopathy, with characteristic histologic features. Treatment is repeated phlebotomy and family screening is advised.
Sarcoidosis
Sarcoidosis is a systemic disease resulting in the formation of noncaseating granulomas that can infiltrate the myocardium. Sarcoidosis is associated with restrictive cardiomyopathy in 5% of patients and can later progress to dilated cardiomyopathy. Sarcoidosis is difficult to diagnose unless there is other organ involvement (usually pulmonary). Sarcoidosis may be suspected in patients with cardiomyopathy and lymphadenopathy, skin rashes, or splenomegaly. Cardiac sarcoid is associated with ventricular tachycardia and conduction abnormalities (especially complete heart block) that can cause syncope and sudden cardiac death. Endomyocardial biopsy may show findings specific for sarcoidosis but because of the patchy nature of the disease biopsy can miss characteristic lesions, resulting in a low overall sensitivity. Combined myocardial perfusion imaging and FDG PET, as well as cardiac MRI, can sometimes be useful in diagnosing and assessing the degree of active inflammation in patients with suspected cardiac involvement. Cardiac granulomas occasionally respond to steroids but turn to scar tissue. Sudden death is not prevented by steroids. Regular Holter monitoring is recommended to look for evidence of atrioventricular block, which should be treated with permanent pacemaker insertion, or ventricular arrhythmias, which should be treated with an ICD.
Arrhythmogenic Right Ventricular Cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a rare but increasingly recognized condition characterized morphologically by apparent patchy apoptosis of the right and, to a lesser extent, left ventricles. Arrhythmogenic right ventricular cardiomyopathy is sometimes called fat cardiomyopathy because of fatty infiltration of the right ventricle. There is a familial incidence of ARVC in more than 50% of patients, generally with an autosomal dominant mode of inheritance.17
Presentation of ARVC is usually in early adulthood, with symptoms consistent with supraventricular and ventricular arrhythmias or with right-sided heart failure. Family screening may reveal ARVC but often sudden death is the first sign of ARVC with the diagnosis made postmortem. Endomyocardial biopsy is associated with an increased risk of perforation and tamponade. Diagnostic features are summarized in Table 4.
Table 4: Diagnostic features of arrhythmogenic right ventricular cardiomyopathy
| Diagnostic Modality | Features |
|---|---|
| Electrocardiography | Epsilon waves (slurred ST segments) V1-3, inverted T waves V2, V3*) in absence of right bundle branch block |
| Echocardiography | Localized RV aneurysm, isolated RV failure |
| Magnetic Resonance Imaging | Fatty infiltration of right ventricle |
| Histology | Fatty infiltration of right ventricle |
RV = right ventricular.
* In patients older than 12 years.
© 2003 The Cleveland Clinic Foundation.
Managing these patients with ARVC is difficult and controversial. Control and prevention of potentially lethal ventricular arrhythmias are of paramount importance and have been approached with antiarrhythmic medications, radiofrequency ablation and, inevitably, ICDs. Control of right heart failure is difficult and sometimes impossible by conventional therapy. Cardiac transplantation provides effective therapy in selected cases.
Unclassified Cardiomyopathies
Noncompacted Myocardium
Noncompaction of the myocardium manifests in early childhood. Noncompacted myocardium appears to occur as a result of an arrest in endomyocardial morphogenesis and is often associated with other cardiac abnormalities and extracardiac anomalies. Patchy preservation of the spongy morphology of the embryonic heart occurs, with persisting myocardial sinusoids and prominent ventricular trabeculations seen with echocardiography. Noncompaction of the myocardium involves the right ventricle and is occasionally biventricular. Clinical presentation includes ventricular arrhythmias, thromboembolism, and progressive systolic or diastolic heart failure, or both. Noncompacted myocardium is a malignant condition with a high mortality usually occurring in childhood. Patients are treated conventionally and some have successfully undergone heart transplantation.
Systolic Dysfunction with Minimal Dilation
Systolic dysfunction with minimal dilation, as its name suggests, is characterized by systolic dysfunction, an ejection fraction lower than 30% (with no evidence of restrictive physiology by definition) and preserved left ventricular dimensions. Histologically, there is little myofibrillar loss. Systolic dysfunction with minimal dilation carries a poor prognosis. Patients are treated with conventional approaches. A family history of dilated cardiomyopathy is not uncommon.
Mitochondrial Disease
Mitochondrial cardiomyopathy, arising from mutations in mitochondrial DNA, with resultant impaired oxidative phosphorylation, is transmitted through the maternal line. The resultant cardiomyopathy is characterized by progressive hypertrophy, dilation, and arrhythmias. Mitochondrial diseases generally are systemic, in tissues with high metabolic activity, and give rise to syndromes. The MELAS (mitochondrial encephalopathy, lactic acidosis, and strokelike) syndrome can manifest as cardiomyopathy. When a mitochondrial myopathy is suspected, electron microscopy of EMB specimens may reveal giant mitochondria, concentric cristae, and intramitochondrial inclusions. However, skeletal muscle biopsy should be considered first, because it is a safer alternative.
Endocardial Fibroelastosis
Endocardial fibroelastosis is a rare condition that usually manifests in infancy or early childhood and is characterized by thickening of the left ventricle and left-sided cardiac valves. Multiple modes of inheritance have been described. Dilated or restrictive cardiomyopathy can result.
General Consideration
Signs and Symptoms
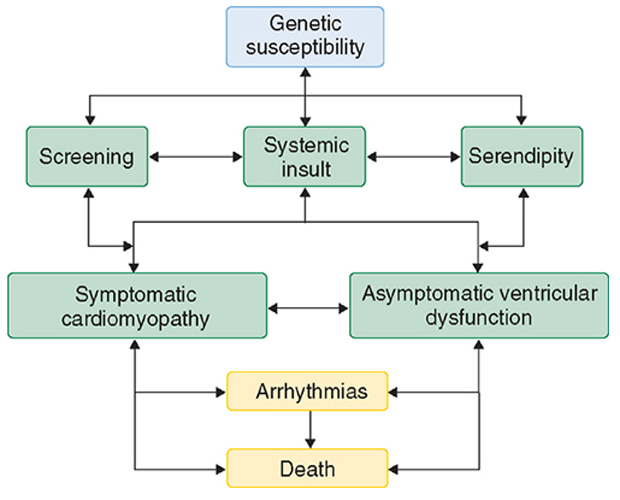
The cardiomyopathies represent a spectrum of disease and as such have various presentations (Figure 1). Patients can remain asymptomatic with cardiomyopathy diagnosed only by screening or postmortem examination. Patients may present with symptoms of heart failure (see Heart Failure), chest pain, or dysrhythmias. The clinical course can fluctuate. Unfortunately, the most common clinical presentation is one of progressive deterioration, with worsening heart failure and death occurring over a variable time course.
Diagnosis
A careful history is essential, with particular emphasis on family history. A family tree should be constructed to ascertain whether a pedigree exists that is consistent with familial cardiomyopathy. A family history can necessitate requesting autopsy reports and medical records, because a high index of suspicion is required. Additional features in the history should focus on exposure to cardiotoxins such as alcohol or cocaine. Specific interrogation about a protracted flulike illness or respiratory tract infection can suggest previous myocarditis.
Key points to assess in patients with known or suspected cardiomyopathy are as follows:
- Establishing the onset and severity of symptoms of dyspnea, fatigue, fluid retention, and effect on activities of daily living
- Conventional risk factors for vascular disease (eg, smoking, hypertension, diabetes, hyperlipidemia) or prior cardiac events (eg, MI, coronary artery bypass grafting)
- Family history of heart disease, especially sudden death
- Alcohol, amphetamine, or cocaine use
- Any past or current major medical illness
The use of genetic screening in the management of cardiomyopathy is under evaluation in many centers, but it has not yet proved to be clinically useful. Genetic testing is currently recommended if a family mutation is known. Genetic testing can also be useful for any patients with confirmed arrhythmogenic right ventricular cardiomyopathy.
Screening first-degree relatives of patients with known or suspected familial cardiomyopathy is currently best achieved by physical examination, ECG, and echocardiography. The age at which screening should commence and how often it should be continued are unclear.
Examination should focus on whether the heart is palpably dilated, the presence of murmurs, and additional heart sounds (gallops). It is important to look beyond the cardiovascular system to consider a possible systemic disorder that may be contributory or causal, such as hemochromatosis or thyrotoxicosis.
There are five useful clinical signs to establish the presence and severity of cardiomyopathy: general appearance (cachexia and dyspnea at rest indicate severe impairment), hypotension, tachycardia, elevated jugular venous pressure, and displaced left ventricular point of maximal impulse (PMI).
Basic investigations should include a chest radiograph, ECG, and echocardiogram. Screening laboratory investigations include a complete blood cell count and renal, glucose, lipid, liver, and thyroid panels. It is reasonable to measure ferritin levels if hemochromatosis is suspected. The usefulness of viral titers has not been proven, although it may be reasonable to perform specific viral serology, such as for HIV, if indicated by the history.

B-type natriuretic peptide (BNP) has been identified as a useful marker for the diagnosis, severity, and prognosis in patients with heart failure. B-type natriuretic peptide levels correlate with functional class but not with ejection fraction.
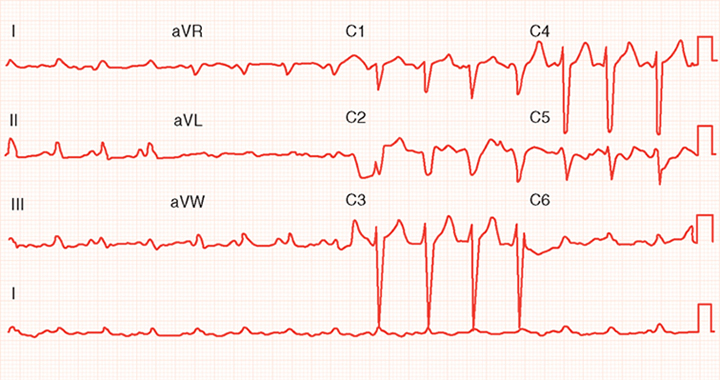
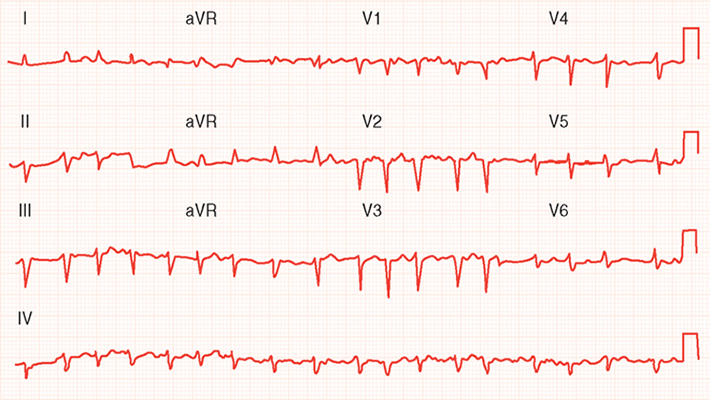
The five most common abnormalities seen on the ECG are Q waves (from a previous MI), diffuse ST-segment abnormalities, left bundle branch block (or any intraventricular conduction delay), atrial fibrillation, and abnormal P waves (biphasic in leads V1 and V2 [left atrial overload]). Figure 2 shows intraventricular delay and diffuse ST-segment abnormalities in an ECG from a patient with dilated cardiomyopathy. Figure 3 shows atrial fibrillation, poor R wave progression, and diffuse ST-segment abnormalities in an ECG from a patient with ischemic cardiomyopathy.
Common abnormalities seen on the chest radiograph include cardiomegaly, interstitial edema, pleural effusion(s), and evidence of previous sternotomy (sternal wires).
Common abnormalities seen on the echocardiogram include increased chamber dimensions, reduced fractional shortening or ejection fraction, functional mitral and tricuspid valvular regurgitation, regional wall motion abnormalities, and myocardial thickening (hypertrophy or infiltration).
Common abnormalities seen on Holter monitoring include premature ventricular complexes, premature atrial complexes, atrial fibrillation (either sustained or paroxysmal), nonsustained ventricular tachycardia, and first- or second-degree atrioventricular block.
It is important to rule out ischemia as a cause of ventricular dysfunction. This can most definitively be determined by cardiac catheterization and coronary arteriography. Noninvasive modalities such as dobutamine stress echocardiography and nuclear stress testing are available at most centers as screening tests for coronary artery disease. Positron emission tomography and MRI are other noninvasive modalities that are only available at large centers.
Role of Endomyocardial Biopsy in Diagnosis of Cardiomyopathy
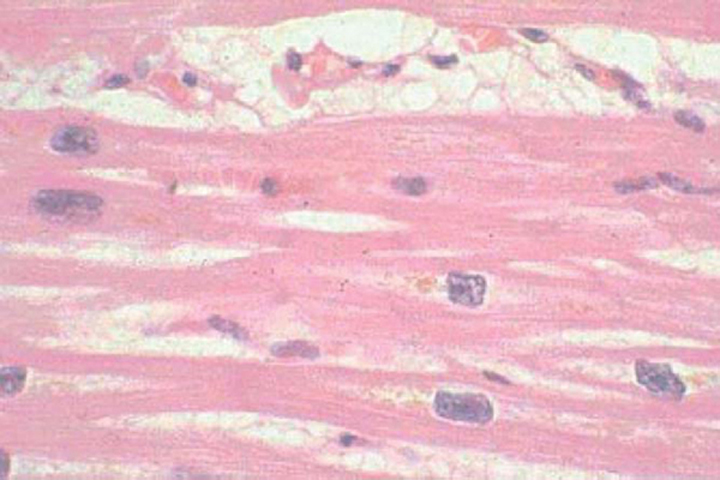
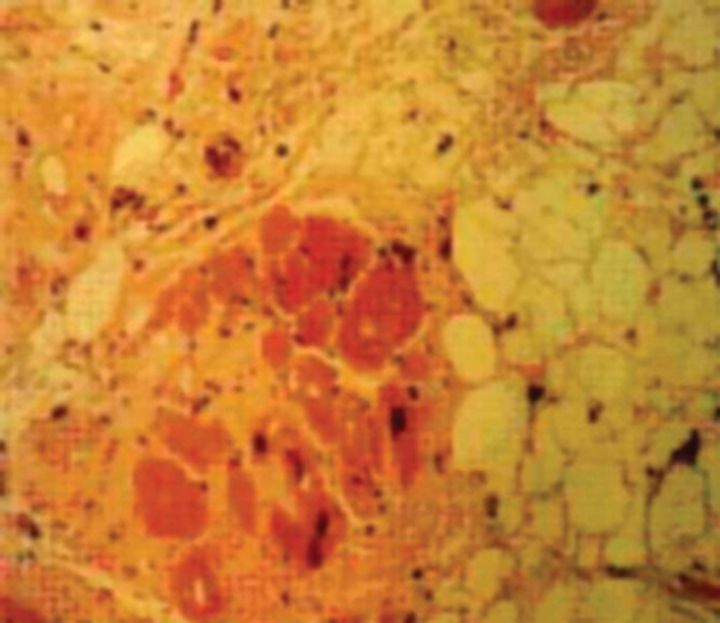
Endomyocardial biopsy is not indicated in the routine evaluation of cardiomyopathy,2 even though it has a complication rate of less than 1%.15 Most histologic specimens demonstrate nonspecific changes of myocyte hypertrophy, cell loss, and fibrosis (Figure 4) and do not affect medical management. Histologically specific changes do occur in sarcoid (although they are patchy, reducing sensitivity), amyloid, hemochromatosis (Figure 5), endocardial fibroelastosis, Löffler’s endocarditis, and ARVC (Figure 6). These conditions may be diagnosed with other less invasive tests. In addition, there are no data proving that treatment improves outcome in cardiac sarcoid or amyloid, although some support the use of EMB in suspected cases of amyloid to establish the diagnosis and predict a poor prognosis (mean survival, 6 to 12 months). Because of recurrent disease, cardiac amyloid generally renders the patient ineligible for cardiac transplantation.
Endomyocardial biopsy in acute myocarditis is not useful because aggressive immunosuppressive regimens, once believed to be efficacious, do not appear to improve outcome. However, the one exception is that giant cell myocarditis, suggested by a rapidly progressive and downhill course, may benefit from aggressive therapy (eg, intensive hemodynamically guided heart failure therapy, immunosuppression, left ventricular assist device, transplantation). In addition to its use for potential giant cell myocarditis, EMB, combined with cardiac imaging techniques, may be used to document anthracycline toxicity, although BNP level may be a more sensitive marker.
Endomyocardial biopsy is sometimes useful for distinguishing constrictive from restrictive pathology (the latter being associated with infiltration on EMB).19 However, this distinction can generally be made with a number of imaging techniques, including echocardiography with diastolic studies (see Pericardial Disease), MRI, computed tomography, and cardiac catheterization.
Treatment and Outcomes of Cardiomyopathies
Therapy for cardiomyopathy generally is the same as for heart failure. However, the usefulness of therapies for specific populations remains to be defined, including those for patients with asymptomatic left ventricular dilation. Neither steroids nor intravenous immunoglobulin are useful in the management of cardiomyopathy.
In the absence of a specific remediable cause (eg, peripartum, alcoholic, ischemic, or hibernating cardiomyopathy), the overall outcome is poor. The 5-year survival rate of patients with diagnosed heart failure is 50%.20 This is paralleled by a high morbidity, characterized by polypharmacy and multiple hospital admissions. Several clinical and laboratory features imply a poor prognosis (Table 5).13,21,22
Table 5: Predictors of Poor Outcome in Dilated Cardiomyopathy
| Test | Features |
|---|---|
| Clinical findings | Increased age, male gender, ischemic heart disease, diabetes, syncope, right heart failure symptoms, symptomatic ventricular arrhythmias, persistent gallop rhythm, persistent jugular venous distention, systemic hypotension, peripheral vascular disease |
| Laboratory findings | Hyponatremia, persistently elevated B-type natriuretic peptide and A-type natriuretic peptide levels, elevated norepinephrine and renin levels |
| Electrocardiography | Left bundle branch block, first- and second-degree atrioventricular blocks |
| Echocardiography | Increased ventricular dimensions, reduced ejection fraction, restrictive diastolic filling pattern, severe mitral or tricuspid regurgitation, or both |
| Chest radiography | Increased cardiothoracic ratio |
| Coronary angiography | Multivessel obstructive disease |
| Hemodynamic data | Pulmonary capillary wedge pressure >20 mmHg, cardiac index <2.5 L/min/m2, pulmonary hypertension, elevated central venous pressure |
| Cardiopulmonary exercise test | Maximal systemic oxygen uptake <12 mL/kg/min |
| Endomyocardial biopsy | Loss of intracellular myofilaments |
Data from Topol E: Textbook of Cardiovascular Medicine. Philadelphia: Lippincott Williams & Wilkins, 2002; Bart BA, Shaw LK, McCants CB, Jr., et al. Clinical determinants of mortality in patients with angiographically diagnosed ischemic or nonischemic cardiomyopathy. J Am Coll Cardiol 1997;30:1002-1008; and Koelling TM, Aaronson KD, Cody RJ, et al. Prognostic significance of mitral regurgitation and tricuspid regurgitation in patients with left ventricular systolic dysfunction. Am Heart J 2002; 144:524–529.
Metabolic stress testing is useful to gauge effort tolerance objectively, and a peak VO2 uptake of less than 14 mL/kg/min is generally accepted as a criterion for heart transplantation. Heart transplantation provides a median survival of 12 years and is effective palliation for appropriately selected patients.
“Which patients should I refer to a cardiologist?” Because of the poor prognosis in most cases of cardiomyopathy, all patients for whom active treatment is contemplated should be referred to a cardiologist to ascertain the cause so that an aggressive, tailored etreatment plan can be initiated.
Summary
- Cardiomyopathies are diseases of the myocardium associated with cardiac dysfunction, often resulting in the clinical syndrome of heart failure.
- Dilated cardiomyopathy is defined as an ejection fraction of lower than 40% in the presence of increased left ventricular dimensions. There are a number of possible causes.
- Restrictive cardiomyopathy is a disease of the myocardium, characterized by restrictive filling and reduced diastolic volume of the ventricles, with normal or near-normal systolic function.
- Cardiomyopathies are diagnosed by history, physical examination, ECG, chest x-ray, echocardiogram and, in some cases, EMB.
References
- Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation 1996; 93:841–842.
- Hunt SA, Abraham WT, Chin MH, et al. American College of Cardiology; American Heart Association Task Force on Practice Guidelines; American College of Chest Physicians; International Society for Heart and Lung Transplantation; Heart Rhythm Society: ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): Developed in collaboration with the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: Endorsed by the Heart Rhythm Society. Circulation 2005; 112:e154–e235.
- Goldstein S, Ali AS, Sabbah H. Ventricular remodeling. Mechanisms and prevention. Cardiol Clin 1998; 16:623–632, vii-viii.
- Arbustini E, Morbini P, Pilotto A, et al. Familial dilated cardiomyopathy: From clinical presentation to molecular genetics. Eur Heart J 2000; 21:1825–1832.
- Griffin BP, ed. Manual of Cardiovascular Medicine, 4th edition. Philadelphia, PA: Lippincott Williams & Williams; 2012.
- Cooper LT Jr. Myocarditis. N Engl J Med 2009; 360:1526–1538.
- Kuhl U, Pauschinger M, Noutsias M, et al. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 2005; 111:887–893.
- Magnani JW, Dec GW: Myocarditis. Current trends in diagnosis and treatment. Circulation 2006; 113:876–890.
- Piano MR: Alcoholic cardiomyopathy. Incidence, clinical characteristics, and pathophysiology. Chest 2002; 121:1638–1650.
- Feldman J, Auer J, Berent R, et al. Cardiovascular complications of cocaine use. N Engl J Med 2001; 345:1575–1576.
- Yeh ETH, Tong AT, Lenihan DJ, et al. Cardiovascular complications of cancer therapy: Diagnosis, pathogenesis, and management. Circulation 2004; 3122–3131.
- Elkayam U, Tummala PP, Rao K, et al. Maternal and fetal outcomes of subsequent pregnancies in women with peripartum cardiomyopathy. N Engl J Med 2001; 344:1567–1571.
- Topol E. Textbook of Cardiovascular Medicine. Philadelphia, Lippincott Willaims & Wilkins, 2002.
- Felker GM, Thompson RE, Hare JM, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med 2000; 342:1077–1084.
- Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med2001; 345:9–16.
- Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation 2005; 112:2047–2060.
- Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: Clinical impact of molecular genetic studies. Circulation 2006; 113:1634–1637.
- Wu LA, Lapeyre AC 3rd, Cooper LT. Current role of endomyocardial biopsy in the management of dilated cardiomyopathy and myocarditis. Mayo Clin Proc 2001; 76:1030–1038.
- Remme WJ, Swedberg K. Guidelines for the diagnosis and treatment of chronic heart failure. Eur Heart J 2001; 22:1527–1560.
- Levy D, Kenchaiah S, Larson MG, et al. Long-term trends in the incidence of and survival with heart failure. N Engl J Med 2002; 347:1397–1402.
- Bart BA, Shaw LK, McCants CB Jr, et al. Clinical determinants of mortality in patients with angiographically diagnosed ischemic or nonischemic cardiomyopathy. J Am Coll Cardiol 1997;30:1002–1008.
- Koelling TM, Aaronson KD, Cody RJ, et al. Prognostic significance of mitral regurgitation and tricuspid regurgitation in patients with left ventricular systolic dysfunction. Am Heart J 2002; 144:524–529.