
Myeloproliferative Neoplasms
Michael Keng
Anjali Advani
Karl Theil
Published: December 2013
The myeloproliferative neoplasms (MPNs), previously termed the myeloproliferative disorders, are characterized by the clonal proliferation of one or more hematopoietic cell lineages, predominantly in the bone marrow, but sometimes in the liver and spleen.1 In contrast to myelodysplastic syndromes (MDS), MPNs demonstrate terminal myeloid cell expansion into the peripheral blood. In the 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms, MPNs include: chronic myelogenous leukemia (CML), chronic neutrophilic leukemia, polycythemia vera (PV), primary myelofibrosis (PMF), essential thrombocythemia (ET), chronic eosinophilic leukemia, mastocytosis, and unclassifiable MPNs.2Overlap disorders (MDS/ MPN) are those chronic myeloid disorders unable to be classified as “classic” MPN or MDS. These include: chronic myelomonocytic leukemia (CMML), atypical CML, juvenile myelomonocytic leukemia, and unclassifiable MDS/MPN.2
CML is the only MPN that is characterized by the chromosomal translocation t (9;22), BCR-ABL fusion gene. The most commonly recognized mutation in the remainder of the MPNs is Janus kinase 2 (JAK2) V617F.2 It is present in greater than 90% of patients with PV and approximately half of those with PMF or ET.2 This mutation substitutes phenylalanine for valine at position 617 in the JH2 domain (Val617Phe, V617F) of exon 14, leading to constitutive activation of the JAK-STAT and other pathways resulting in uncontrolled cell growth.3 Subsequent to this, additional mutations within exon 12 of JAK2 have been identified in PV.4 JAK2 allele burden might be important in identifying high-risk patients with PV or ET (i.e., those at risk for requiring treatment with chemotherapy or those at risk for developing major cardiovascular complications).5 Mutations within exon 10 of MPL, the thrombopoietin receptor, have also been identified in ET and PMF.6 The presence of these mutations is determined by polymerase chain reaction (PCR) assays and may be helpful in differentiating a MPN from a reactive cause for elevated counts.
This chapter reviews the definition, prevalence, pathophysiology, signs and symptoms, diagnosis, treatment, and outcomes of PV, PMF, and ET. CML and CMML are discussed in the “Chronic Leukemias” section.
Polycythemia Vera
Definition and Etiology
PV is a clonal disorder characterized by the overproduction of mature red blood cells in the bone marrow.1Myeloid and megakaryocytic elements are also often increased. No obvious cause exists.7 Genetic and environmental factors have been implicated in rare cases. Familial PV has been associated with mutation of the erythropoietin (EPO) receptor.8 An increased number of cases have been reported in survivors of the atomic bomb explosion in Hiroshima during World War II.
Epidemiology
The disorder typically occurs in the sixth or seventh decade of life. The prevalence of the disease is approximately 5 per million population; it occurs more commonly in men and in men and women of East European Jewish ancestry.1,4
Pathophysiology
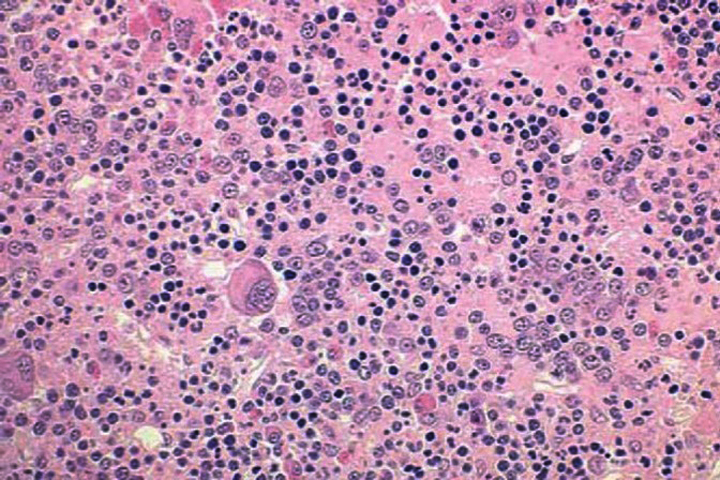
The primary defect involves a pluripotent stem cell capable of differentiating into red blood cells, granulocytes, and platelets.7Clonality has been demonstrated through glucose-6-phosphate dehydrogenase studies as well as restriction fragment length polymorphism of the active X chromosome.8 Erythroid precursors in PV are exquisitely sensitive to erythropoietin, which leads to increased red blood cell production. Precursors in PV are also more responsive to cytokines such as interleukin-3 (IL-3), granulocyte-macrophage colony-stimulating factor, and steel factor. Myeloid and megakaryocytic elements are often increased in the bone marrow (Figure 1). More than 60% of patients have endogenous megakaryocyte colony unit formation. The abnormal proliferation of PV is due to constitutive activation of the JAK-STAT pathway, with the majority of patients (>90%) harboring the V617F mutation.2 A similar JAK2 exon 12 mutation is found in the few patients lacking the V617F mutation.2
Increased red blood cell production in PV leads to an increased red cell mass and increased blood viscosity. This in turn can lead to arterial or venous thrombosis, bleeding, or both.1 The hematocrit is directly proportional to the number of thrombotic events.7 Investigators have demonstrated a reduction in cerebral blood flow in patients with hematocrits between 53% and 62%.8 An increased platelet count can also contribute to bleeding and thrombosis. Although platelet aggregation abnormalities exist in most patients, these abnormalities do not appear to correlate with the risk of bleeding or thrombosis. Increased production and breakdown of blood cells can lead to hyperuricemia and hypermetabolism.
Signs and Symptoms
Patients may be asymptomatic at the time of diagnosis and have only isolated splenomegaly, erythrocytosis, or thrombocytosis.8 However, most patients develop symptoms as the hematocrit, platelet count, or both increase. An elevated white blood cell (WBC) count is found in 50% to 60% of patients. Symptoms of hyperviscosity associated with an elevated hematocrit include headache, blurred vision, and plethora.1
Thrombosis in small blood vessels can lead to cyanosis, erythromelalgia (painful vessel dilation in the extremities), ulceration, or gangrene in the fingers or toes. Thrombosis in larger vessels can lead to myocardial infarction, deep venous thrombosis, transient ischemic attacks, and stroke. A cerebrovascular event precedes the diagnosis in 35% of patients with PV.7 Unusual sites of thromboses also tend to be seen more frequently in PV—splenic, hepatic, portal, and mesenteric.
Of patients with Budd-Chiari syndrome (hepatic–inferior vena cava obstruction), 10% have coexisting PV. Abnormalities in platelet function lead to epistaxis, bruising, and gastrointestinal and gingival bleeding in 2% to 10% of patients. Severe bleeding episodes are unusual. Hypermetabolism caused by increased blood cell turnover can lead to hyperuricemia, gout, stomach ulcers, weight loss, and kidney stones. Pruritis is especially common after a warm bath or shower. As the disease progresses, many patients develop abdominal pain secondary to hepatomegaly, splenomegaly, or both.
Diagnosis
PV should be suspected in men with a hemoglobin greater than 18.5 g/dL and in women with a hemoglobin greater than 16.5 g/dL.2 An elevated red cell mass, measured using direct tagging of red blood cells with chromium 51, was previously important in making the diagnosis.2 However, this is rarely if ever used now. The presence of the JAK2 mutation is now a major criterion for diagnosis.
Secondary causes of polycythemia must be ruled out. Many conditions can physiologically increase the production of EPO. EPO overproduction occurs in: hypoxia (e.g., pulmonary disease, high altitude, smoking due to carboxyhemoglobin, cyanotic cardiac disease, and methemoglobinemia), tumors (e.g., kidney, brain, hepatoma, uterine fibroid, and pheochromocytoma), renal artery stenosis, and renal cysts. Other causes include androgen therapy, congenital erythrocytosis, EPO receptor hypersensitivity, auto-transfusion (blood doping), and self-injection of EPO. Serum EPO levels should be low to normal in patients with PV but high in patients with secondary polycythemia, although there may be some overlap. Molecular testing for the JAK2 V617F or other functionally similar mutation currently plays a central role in the diagnosis of PV as a way of separating neoplastic from reactive myeloid proliferations.
The initial diagnostic criteria defined by the Polycythemia Vera Study Group (PVSG) have undergone changes over the last several years. The current WHO criteria requires the presence of both major criteria and one minor criterion or the presence of the first major criterion together with two minor criteria to make a diagnosis of PV.2
Polycythemia Vera Study Group Criteria9
A Criteria
Elevated red cell mass more than 25% above mean normal predicted value, or hemoglobin higher than 18.5 g/dL in men or 16.5 g/dL in women, or higher than the 99th percentile of method-specific reference range for age, sex, and altitude of residence
No cause of secondary erythrocytosis, including:
- Absence of familial erythrocytosis
- No elevation of EPO caused by:
- Hypoxia (arterial PO2 <92%)
- High oxygen affinity hemoglobin
- Truncated EPO receptor
- Inappropriate EPO production by tumor
Splenomegaly
Clonal genetic abnormality other than the Philadelphia chromosome or BCR/ABL1 fusion gene in marrow cells
Endogenous erythroid colony formation in vitro
B Criteria
Thrombocytosis higher than 400 x 109/L
Leukocytosis higher than 12 x 109/L
Bone marrow biopsy showing panmyelosis with prominent erythroid and megakaryocyte proliferation
Low serum EPO levels
2008 WHO Criteria for PV2
Major Criteria
Hemoglobin greater than 18.5 g/dL in men, 16.5 g/dL in women or other evidence of increased red cell volume
- Hemoglobin or hematocrit greater than the 99th percentile of method-specific reference range for age, sex, altitude of residence or hemoglobin greater than 17 g/dL in men, 15 g/dL in women if associated with a documented and sustained increase of at least 2 g/dL from a patient’s baseline value that cannot be attributed to correction of iron deficiency, or
- Elevated red cell mass greater than 25% above mean normal predicted value
Presence of JAK2 V617F or other functionally similar mutation such as JAK2 exon 12 mutation
Minor Criteria
Bone marrow biopsy showing hypercellularity for age with trilineage growth (panmyelosis) with prominent erythroid, granulocytic, and megakaryocytic proliferation
Serum EPO level below the reference range for normal
Endogenous erythroid colony formation in vitro
Treatment
Treatment of PV focuses on decreasing the hemoglobin level, thereby reducing plasma viscosity and its attendant complications. Therapeutic options include phlebotomy, radioactive phosphorus (32P), and myelosuppressive agents. The goal of therapy is a hematocrit of 45% for men and 42% for women on the basis of cerebral blood flow studies.7 Several clinical trials have tried to address the optimal treatment of PV.
Treatment for PV should be risk-adapted.10 Patients at high risk for thrombosis include patients older than 60 years and those with a prior history of thrombosis. Low-risk patients include those who are younger than 60 years with no history of thrombosis, a platelet count below 1,500 x 109/L, and the absence of cardiovascular risk factors (e.g., smoking, hypertension, congestive heart failure).10 In the PSVG-01 study, thrombotic events were increased in the phlebotomy arm, particularly in patients with a history of thrombosis, advanced age, or high phlebotomy requirement.7 Therefore, high-risk patients should be treated with phlebotomy plus cytoreductive therapy or interferon. Hydroxyurea is typically used as first-line therapy.7 However, interferon should be used in women of childbearing age and in patients who cannot tolerate hydroxyurea.7 Low or intermediate-risk patients may be treated with phlebotomy alone.
In the PVSG-01 study, there was an increased risk of leukemia in the 32P and chlorambucil arms (two or three times that seen in the phlebotomy arm).7 Because of the increased leukemogenicity associated with chlorambucil, hydroxyurea, which inhibits ribonucleotide reductase, is now the most widely used myelosuppressive agent. Side effects of hydroxyurea include myelosuppression, macrocytosis, leg ulcers, increased creatinine level, and jaundice.8 A recent large study has demonstrated no increased incidence of leukemia in PV patients treated with hydroxyurea.10 For older, high-risk patients, 32P and busulfan can be used to help with issues of compliance and convenience, especially if the patient’s life expectancy is less than 10 years; however, these are rarely used now.
Myelosuppressive agents should also be used for symptomatic splenomegaly or pruritis intractable to antihistamines.7 Interferon-alfa may also be used in the place of hydroxyurea for myelosuppression, particularly in younger patients and in patients with intractable pruritis. Side effects of interferon include flu-like syndrome, fevers, neuritis, and fatigue8; although the pegylated versions of interferon are better tolerated. Patients with PV who are undergoing surgery are at extremely high risk of developing postoperative complications if their erythrocytosis is not controlled before surgery.
Patients with PV and no drug contraindications or evidence of acquired von Willebrand syndrome should be treated with low-dose aspirin.10 One study (ECLAP) has demonstrated an antithrombotic benefit for low-dose aspirin (100 mg/day) in patients already receiving treatment for PV.10 Patients with erythromelalgia also experience a rapid relief of their symptoms after low-dose aspirin.7
JAK2 inhibitors are being evaluated in clinical trials for PV; but are not currently approved for this indication.
Regardless of the therapy used, a recent randomized clinical trial confirmed that a hematocrit goal of less than 45% is the target of PV therapy.11 This goal is associated with a four-fold decrease of major cardiovascular events and prevention of thrombotic complications.11
Outcomes
The median survival is more than 10 years with treatment. The major causes of death in untreated patients are thrombosis and hemorrhage.1 Fewer than 10% of patients develop acute myeloid leukemia.1 Fifteen percent of patients develop postpolycythemic myelofibrosis (MF) at an average interval of 10 years from diagnosis.7Once patients develop MF, most patients die within 3 years—as MF often transforms to acute myeloid leukemia.7
Primary Myelofibrosis
Definition and Causes
PMF, previously called chronic idiopathic myelofibrosis (CIMF) in the 2001 Who classification, is also known as agnogenic myeloid metaplasia.1,12 In PMF, a clonal hematopoietic stem cell expansion in the bone marrow is accompanied by a reactive nonclonal fibroblastic proliferation and marrow fibrosis. As the bone marrow becomes fibrotic and normal hematopoiesis can no longer occur, extramedullary hematopoiesis (myeloid metaplasia) occurs in the liver and spleen.13 The cause is unknown.
Epidemiology
The incidence of PMF is 0.5 to 1.5 per 100,000 population.1 The risk of developing PMF is increased by exposure to benzene or radiation (higher incidence of individuals exposed to radiation in Hiroshima). The disease is more common in Caucasians, and the median age at diagnosis is 67 years.8 Men and women are affected equally. As noted earlier, patients with PV and other MPNs can develop secondary MF late in the course of their disease.7
Pathophysiology
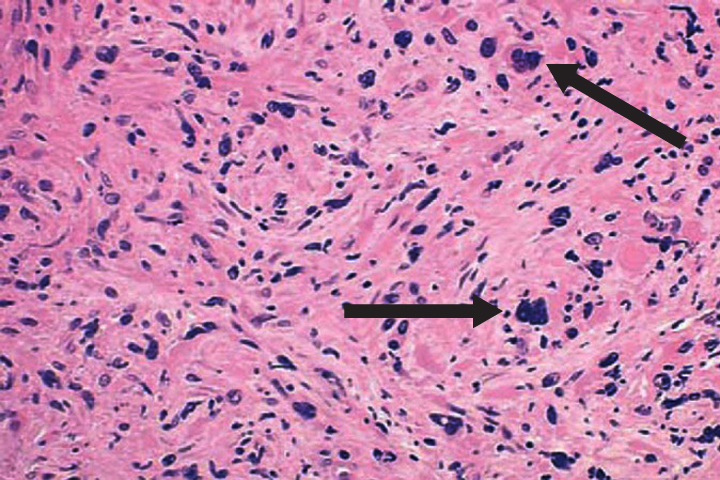
PMF is described by marrow fibrosis and extramedullary hematopoiesis. Clonal studies have demonstrated a stem cell origin.13 The clonal proliferation of hematopoietic stem cells produces growth factors (platelet-derived growth factor, transforming growth factor-B, epidermal growth factor, and basic fibroblastic growth factor) leading to fibrosis of the marrow.1,8 Initially, the bone marrow is hypercellular, but normal hematopoiesis is diminished as the bone marrow becomes fibrotic and patients become pancytopenic (Figure 2).1 Because of this, extramedullary hematopoiesis occurs in the liver and spleen causing these organs to enlarge.
Signs and Symptoms
Many symptoms are attributable to the pancytopenia associated with PMF. Pancytopenia occurs as a result of decreased hematopoiesis and splenic sequestration. Most patients are anemic and feel short of breath and fatigued. Thrombocytopenia and neutropenia can lead to hemorrhage and infection, respectively.
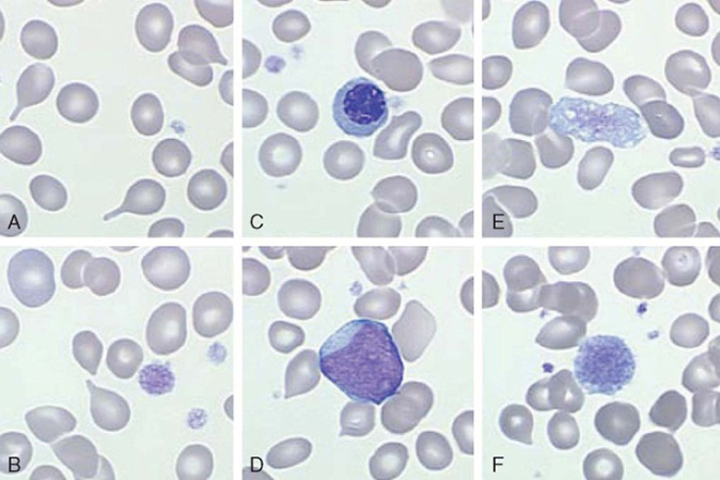
At diagnosis, up to one-third of patients can be asymptomatic. However, common constitutional symptoms include anorexia, weight loss, and night sweats.1 The WBC and platelet counts might increase initially but typically decrease as the disease progresses. The blood film displays a characteristic leukoerythroblastic picture (teardrop poikilocytosis, nucleated red blood cells, and immature myeloid elements) caused by crowding out of normal hematopoietic elements by fibrosis in the bone marrow (Figure 3).8
Patients might note abdominal discomfort and decreased appetite because of splenic and hepatic enlargement resulting from extramedullary hematopoiesis.1 Portal hypertension and jaundice can occur as a result of increased hepatic blood flow.8 Rarely, extramedullary hematopoiesis can occur at other sites, such as the skin, lungs, bladder, genitourinary tract, gastrointestinal tract, and central nervous system. Severe bone pain typically heralds a poor prognosis and often represents a conversion of PMF to acute leukemia.
Diagnosis
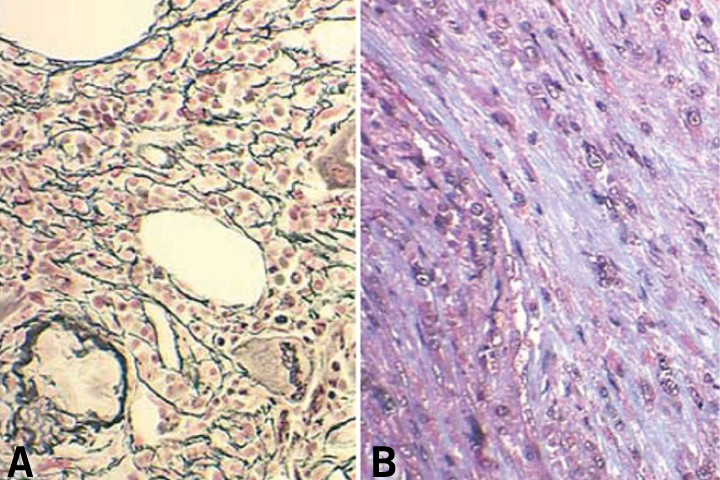
The peripheral blood smear demonstrates a characteristic leukoerythroblastic picture.8 Attempts to perform a bone marrow aspirate and biopsy are often complicated by a dry tap.1 Special stains of the bone marrow biopsy demonstrate increased fibrosis. Stains that contain silver can be used to identify reticulin, the glycoprotein coating of stromal cell strands that appears as black fibers.13 Trichrome stains identify mature collagen as bluish-green fibers, depending on the stain used (Figure 4). The bone marrow biopsy is typically hypercellular initially, and increased numbers of abnormal megakaryocytes with a tendency to form loose to tight clusters are often present.1,8 Cytogenetic abnormalities are found in approximately 50% of patients. These abnormalities include trisomy 8, trisomy 9, del(13q), del(20q), and del(12p).14 The JAK2 mutation is found in approximately half of patients, whereas the MPL 515 mutation is only found in approximately 5% of PMF patients.15
Secondary causes of marrow fibrosis (e.g., metastatic breast cancer, lymphoma, lung cancer, infection, autoimmune disorders), as well as other hematologic disorders (e.g., hairy cell leukemia, CML), need to be excluded. In addition, acute panmyelosis with MF and acute megakaryoblastic leukemia need to be ruled out. These entities also manifest with pancytopenia and marrow fibrosis, but patients typically have no splenomegaly, minimal or absent teardrop poikilocytosis, and increased numbers of blasts.
The WHO criteria for PMF emphasize the role of (1) assessing megakaryocyte morphology in bone marrow biopsies, (2) excluding all secondary causes of MF, and (3) demonstrating the presence of JAK2 or other clonal marker (e.g., MPLW 515) when establishing a diagnosis.2 PMF must meet all of the above three major criteria and 2 of the 4 minor criteria (leukoerythroblastosis, increased serum LDH, anemia, and palpable splenomegaly).1
Treatment
The Dynamic International Prognostic Scoring System (DIPSS) includes eight risk factors which impact survival: age >65 years, hemoglobin <8 g/dL, leukocyte count >25 x 109/L, circulating blasts ≥1%, presence of constitutional symptoms (weight loss >10%, night sweats, or fevers for >1 month), presence of unfavorable karyotype, platelet count <100 x 109/L, and red cell transfusion dependence.16 Unfavorable karyotype is defined as complex karyotype or one to two abnormalities that include +8, -7/7q-, -5/5q-, inv (3), i(17q), 12p-, 11q23 rearrangement.16 Each risk factor is 1 point. Risk groups are defined as: low: 0 points, intermediate-1: 1 point, intermediate-2: 2 points, and high: ≥3 points. In a recent study by the Mayo Clinic, the median overall survival from the time of referral was 20, 14.3, 5.3, and 1.7 years, respectively, for the risk groups above.17
There are no treatments that can completely reverse the process of PMF short of bone marrow transplantation.1 Because of the older median age at diagnosis, many patients are not suitable bone marrow transplant candidates. One transplant study demonstrated a 47% overall survival at 5 years, with regression of fibrosis in 40% of patients.13 The increasing use of nonmyeloablative transplants should extend this option to older patients with PMF. Patients with intermediate-2 or high-risk PMF should be considered for bone marrow transplant.18 Patients <45 years of age should be considered for conventional-intensity conditioning regimens, while reduced intensity (or, nonmyeloablative) transplant should be considered for those between 45 and 65 years of age.18 Patients above age 65 years should be considered for investigational drug therapy.
The role and benefit of using chemotherapeutic agents early in the disease to reverse the fibrosis are highly controversial, and randomized clinical trials are needed to address this issue.8 Most care is directed toward symptomatic management with transfusions (red blood cells, platelets) and growth factors (EPO for anemia, granulocyte-colony stimulating factor for neutropenia). Because of the frequency of red blood cell transfusions, patients can require iron chelation therapy to decrease the risk of iron overload. Androgens (danazol) and, occasionally, low-dose steroids (e.g., prednisone) may also be helpful in managing the anemia associated with ineffective erythropoiesis.
The immunomodulatory drugs thalidomide, lenalidomide (Revlimid), and pomalidomide have all been evaluated in PMF because of their anti-angiogenic, anti-TNF alpha, and T-cell modulating effects.19 Thalidomide was the first drug available. A pooled analysis of small phase 2 studies with thalidomide published between 2000 and 2002 demonstrated a response (increase in hemoglobin, or reduction or elimination of blood transfusion requirements) in 29% of patients with moderate or severe anemia receiving thalidomide at a dosage of 200 to 800 mg/day.20 However, a large number of patients stopped drug secondary to side effects (neuropathy, somnolence). More recent studies have evaluated the first-generation immunomodulatory agent, lenalidomide. Lenalidomide has been evaluated in two separate, but similarly designed phase 2 studies. Overall response rates were 22% for improvement of anemia, 33% for reduction of splenomegaly, and 50% for improvement of thrombocytopenia.21 A subset of patients had an impressive improvement in their anemia or resolution of bone marrow abnormalities (fibrosis, angiogenesis), or both.21 However, the drug was associated with significant myelosuppression. Pomalidomide is 20,000-times more potent than thalidomide in inhibition of TNF-alpha and is not cross-resistant with the other IMIDS.22 Overall anemia response rates of 27% have been demonstrated; however, this response rate was significantly increased (53%) in patients with a spleen <10 cm, peripheral blasts <5%, and the presence of the JAK2 mutation.22 Similar to lenalidomide, myelosuppression is a common toxicity. A Phase 3 trial of pomalidomide versus placebo was recently completed in PMF; and the results of this study are awaited. Corticosteroids increase the activity of all of the immunomodulatory agents through the activation of dual apoptotic signaling.22 The immunomodulatory agents are typically not optimal agents for treating symptomatic splenomegaly associated with PMF; and therapeutic approaches for these patients are discussed below. Treatment with hypomethylating agents in PMF is currently under investigation.
Splenomegaly can require treatment with myelosuppressive agents, splenectomy, or palliative radiation.1 Splenic irradiation is typically associated with transient responses and should be considered for patients too ill for splenectomy or chemotherapy. Splenectomy may be considered for symptomatic splenomegaly; although things have changed with the advent of the JAK2 inhibitors. In a series of 223 patients at the Mayo Clinic, patients experienced durable remissions in constitutional symptoms (67%), transfusion-dependent anemia (23%), portal hypertension (50%), and severe thrombocytopenia (0%) after splenectomy.23 The operative mortality rate was 9% and the morbidity rate was 31%, including postoperative thrombotic complications. Sixteen percent of patients developed hepatomegaly, and 22% of patients developed thrombocytosis, which was associated with an increased risk of perioperative thrombosis.23 Blastic transformation was 16.3% in this series, and the risk was increased in patients with splenomegaly and preoperative thrombocytopenia, suggesting that pre-splenectomy thrombocytopenia may be a surrogate marker of advanced disease.
Ruxolitinib (Jakafi, INCBO18424), a selective JAK1/JAK2 inhibitor, decreases splenomegaly (≥35% reduction in splenic volume in 28% to 42% of patients), ameliorates constitutional symptoms, and improves overall survival in PMF.24 Due to the positive findings from the COMFORT-1 and COMFORT-II phase III trials, ruxolitinib was FDA approved for patients with intermediate or high-risk PMF. Anemia and thrombocytopenia are the most common toxicities.24 Other JAK inhibitors are currently being evaluated in clinical trials. In the future, the clinical development of antifibrotic and antiangiogenesis therapies might play an important role in the therapeutic armamentarium.13
Outcomes
Patients asymptomatic at the time of diagnosis can have an indolent clinical course for several years. However, among the MPNs, PMF has the worst prognosis, with a median survival of 3.5 to 5.5 years.13 The most common causes of death include infection, cardiovascular disease, cerebrovascular disease, hemorrhage or thrombosis, and acute leukemia (10% to 25%). Patients with acute leukemia arising from PMF rarely achieve a remission from induction chemotherapy.8
Essential Thrombocythemia
Definition and Etiology
ET, previously called hemorrhagic thrombocythemia, was first described by Epstein and Goedel in 1934. ET is characterized by a sustained clonal proliferation of megakaryocytes in the bone marrow8, with a peripheral blood platelet count greater than 600 x 109/L. This platelet count threshold has been decreased to greater than 450 x 109/L in the most recent WHO classification.2 Causes of reactive thrombocytosis must be excluded. The underlying cause of the disease is unknown.1
Epidemiology
The incidence of the disease (2.5/100,000 population per year in Olmsted County, Minnesota) is the lowest among the chronic MPNs.1,8 There may be a higher prevalence in younger women. The median age at diagnosis is 60 years.
Pathophysiology
The proliferation of megakaryocytes is primarily caused by clonal stem cells, as confirmed by enzyme and genetic analysis.25 Megakaryocyte progenitor cells in ET are hypersensitive to the action of several cytokines, including IL-3 and IL-6, and possibly thrombopoietin.8,25 This leads to increased platelet production. There is controversy regarding spontaneous megakaryocyte formation in ET. The JAK2 mutation is found in 50% to 60% of ET patients.2 Patients lacking mutations in JAK2 may instead demonstrate activating mutations of the thrombopoietin receptor, MPL, MPL W515K or MPL W515L.2
Increased platelet counts in ET are associated with increased thrombotic and hemorrhagic complications. Decreasing platelet counts in ET (to <600 x 109/L) can decrease thrombotic complications.26 High platelet counts (>1,000 x 109/L) are associated with acquired von Willebrand disease resulting from the adsorption of von Willebrand multimers onto platelet membranes.8 A reduction in the platelet count is associated with correction of the defect and cessation of bleeding. Qualitative abnormalities in the platelets themselves are also likely to contribute to the increased risk of thrombotic and hemorrhagic complications in ET, because reactive thrombocytosis is not associated with an increased risk of thrombosis or bleeding, even with high platelet counts. Platelet aggregation studies in ET are often abnormal.
Signs and Symptoms
The clinical signs and symptoms are similar to those of PV. Many patients are asymptomatic at presentation, and their diagnosis is based on an elevated platelet count. However, patients can present with splenomegaly, hepatomegaly, or hemorrhagic or thrombotic episodes.8 Thirteen percent to 37% of patients experience a hemorrhagic event, and 22% to 84% of patients experience a thromboembolic event.8 Constitutional symptoms, such as weight loss, fever, and pruritis, can also occur.
As with PV, thrombotic episodes can occur in the major vessels or microvasculature (see earlier, “Polycythemia Vera: Signs and Symptoms”). Thrombotic episodes occur more often in older patients and in patients with a history of thrombotic events. This increase in thrombotic risk with age has been attributed to the coexistence of vascular disease in older patients. Events tend to occur mainly in the microvasculature.
Women of childbearing age can present with a spontaneous abortion secondary to placental thrombosis, especially in the first trimester.8 Hemorrhage is most common in the gastrointestinal tract.
Diagnosis
ET is described by persistent nonreactive thrombocytosis, a diagnosis of exclusion. The PVSG was the first group to define ET.27 Each patient needed to fulfill the following criteria: (1) platelet count >600 x 109/L; (2) megakaryocytic hyperplasia on bone marrow aspiration and biopsy, (3) absence of the Philadelphia chromosome; (4) absence of infection, inflammation, and other causes for reactive thrombocytosis; (5) normal red blood cell mass or a hemoglobin concentration <13 g/dL; and (6) the presence of stainable iron in a bone marrow aspiration or ≤1 g/dL increase in hemoglobin concentration after a one month trial of oral iron therapy.
Approximately 20 years later, the WHO revised the criteria for ET.2 The diagnosis requires meeting all four criteria: (1) sustained platelet count ≥450 x 109/L (during the work-up period); (2) bone marrow biopsy specimen demonstrating proliferation mainly of the megakaryocytic lineage with increased numbers of enlarged, mature megakaryocytes; no significant increase or left-shift of neutrophil granulopoiesis or erythropoiesis; (3) not meeting WHO criteria for PV, PMF, CML, MDS or other myeloid neoplasm; and (4) demonstration of JAK2 617VF or other clonal marker, or in the absence of a clonal marker, no evidence for reactive thrombocytosis.
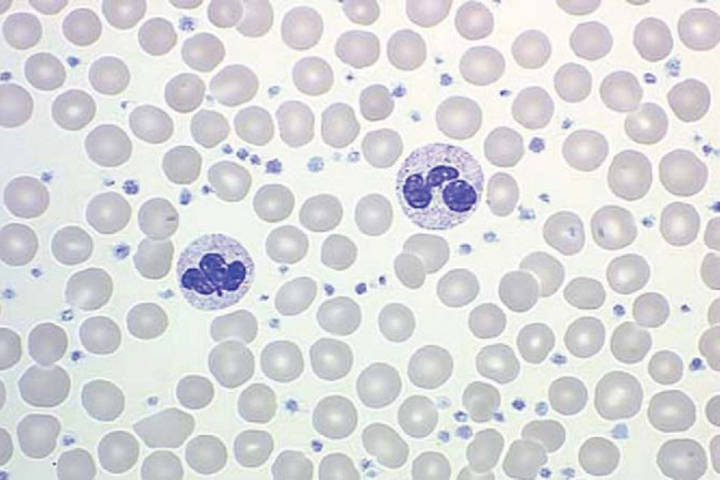
The first step of any patient with thrombocytosis is to examine the peripheral blood film (Figure 5). Automated hematology analyzers can erroneously count platelet-sized particles that are red or white cell fragments as platelets (pseudothrombocytosis).8 Causes of reactive thrombocytosis include: inflammatory states, infection, malignant disease, trauma, blood loss, and the post-splenectomy state.8 JAK2 mutation testing is quite helpful in assisting with diagnosis. Other studies to obtain include: iron studies to rule out iron deficiency; C-reactive protein, erythrocyte sedimentation rate, and fibrinogen to rule out inflammatory states; peripheral blood smear to rule out pseudothrombocytosis; and FISH or PCR for BCR/ABL to rule out CML.28
In one series of 280 patients with thrombocytosis at a university hospital, 82% of cases were reactive thrombocytosis.8Some of these patients have platelet counts greater than 1,000 x 109/L, so the degree of thrombocytosis is not always helpful in making a diagnosis. Reactive thrombocytosis is associated with elevated levels of IL-6 or C-reactive protein in 81% of patients, which can help distinguish it from ET.25 Patients with uncomplicated thrombocytosis secondary to a myeloproliferative disorder typically have undetectable IL-6 levels.13
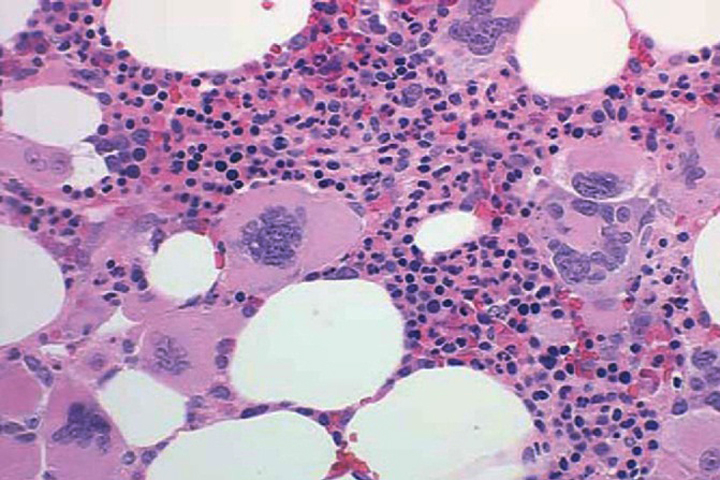
A bone marrow aspirate and biopsy with cytogenetics (and reverse polymerase chain reaction [RT-PCR] for BCR/ ABL1 fusion) can be helpful in excluding CML, which is BCR/ ABL1 positive. Two-thirds of patients with ET have a bone marrow with marked megakaryocytic hyperplasia, morphologically bizarre megakaryocytes with nuclear pleomorphism, and clustering of megakaryocytes (Figure 6).8 Increased myeloid and erythroid precursors, abnormal cytogenetics, minimal reticulin fibrosis, and spontaneous megakaryocyte colony formation may also be present. These features are not typically present in the bone marrow of patients with reactive thrombocytosis. Identifying a JAK2 V617F mutation can be useful in excluding a reactive thrombocytosis. Additional clinical features that suggest ET rather than reactive thrombocytosis include a chronically elevated platelet count, splenomegaly, and history of thrombosis or hemorrhage. An elevated platelet count may also be seen in PV; and the disorder would be considered PV if the other diagnostic criteria are met.2 Finding significant dyserythropoiesis or dysgranulopoiesis should prompt consideration of MDS rather than ET.
Treatment
As with the other MPNs, treatment is based on risk stratification.29 High risk patients include those greater than 60 years of age or those with a history of thrombosis. Low risk patients include those younger than 60 years of age, no history of thrombosis or cardiovascular risk factors, and platelet counts <1,500 x 109/L . Intermediate risk patients comprise the remainder of patients not falling into either of the above groups.
Low-dose aspirin (81 mg) likely decreases the thrombotic risk in ET; although no randomized studies have been conducted as in PV. Patients with a platelet count greater than 1,500 x 109/L and acquired von Willebrand disease should avoid aspirin.8 Asymptomatic patients in the low risk group can be followed with observation only. Patients in the high-risk group should be offered cytoreductive therapy. Controversy exists regarding the treatment of intermediate-risk patients.
A randomized study has demonstrated that hydroxyurea decreases the risk of thrombosis in high-risk patients who have ET from 24% to less than 4% (P = 0.003), compared with no treatment, when the platelet count is decreased to less than 600 × 109/L.26 In addition, maintaining a platelet count less than 400 x 109/L may be associated with a further reduction in thrombosis, although these data have not been confirmed in a randomized trial.25
In addition to hydroxyurea, anagrelide can also control thrombocytosis in most patients.25 The drug works by interfering with megakaryocyte maturation. However, no randomized study has demonstrated that the drug actually decreases the risk of thrombotic events. Therefore, anagrelide should only be used in high-risk patients with ET who do not tolerate or have failed hydroxyurea. Anagrelide’s main side effects include headaches, palpitations, and, rarely, a nonischemic cardiomyopathy.25 These effects are secondary to the drug’s vasodilative inotropic actions; therefore, it should be used cautiously in patients with cardiac disease.25 Anagrelide can also decrease the hematocrit (but not the WBC count). A randomized study (the PT1 trial), has demonstrated that hydroxyurea should be considered first-line treatment for patients with high-risk ET.30 In this study, 809 patients were randomized to receive low-dose aspirin plus either anagrelide or hydroxyurea. The primary composite end point was the actuarial risk of arterial thrombosis, venous thrombosis, serious hemorrhage, or death from hemorrhagic or thrombotic causes.30 After a median follow-up of 39 months, patients in the anagrelide arm were more likely than those in the hydroxyurea arm to have reached the primary end point (odds ratio, 1.57; P = 0.03).
Interferon-alfa is another option for patients, but its use in ET is limited primarily to high-risk women of childbearing age.25 There are no specific recommendations for ET during pregnancy. Although 45% of these women have spontaneous abortions, abortions cannot be predicted by the course of disease, platelet count, or specific treatment. Agents such as 32P or busulfan are rarely used in ET, but can be used in patients whose life expectancy is shorter than 10 years.25
Patients with life-threatening hemorrhagic or thrombotic events should be treated with plateletpheresis in combination with myelosuppressive therapy.25 Patients with a platelet count greater than 1,500 x 109/L and acquired von Willebrand disease should be treated with platelet-reduction therapy and should avoid aspirin.8 These patients also might require treatment with factor VIII concentrates that also contain von Willebrand factor and desmopressin in the setting of a bleeding episode. Finally, patients undergoing surgery have an increased risk of thrombotic and bleeding episodes and should have their platelet counts normalized before surgery.8
Outcomes
Prognosis is highly dependent on the age and history of thrombosis, because thrombosis can be life-threatening for some patients.25 One series has demonstrated that most patients with ET die from thrombotic complications.8 Five- and 10-year survival rates are 74% to 93% and 61% to 84%, respectively. Fewer than 10% of patients with ET convert to acute leukemia, and 5% of patients develop myelofibrosis.8
Summary
- The myeloproliferative neoplasms include polycythemia vera, primary myelofibrosis, essential thrombocythemia, and chronic myeloid leukemia. Chronic myelomonocytic leukemia has features that overlap traditional myelodysplastic syndromes and myeloproliferative neoplasms.
- Myeloproliferative neoplasms are characterized by the clonal proliferation of one or more hematopoietic cell lineages.
- Review of the peripheral blood smear and a bone marrow aspirate/biopsy is needed to make a definitive diagnosis.
- Reverse transcriptase polymerase chain reaction (RT-PCR) or fluorescence in-situ hybridization (FISH) for the BCR/ABL1 gene fusion should be performed to rule out chronic myeloid leukemia.
- Janus kinase 2, a tyrosine kinase, is mutated in a significant proportion of myeloproliferative neoplasms.
- Clinical trials with small molecular inhibitors of JAk2 are currently enrolling patients; and these drugs will likely be an important part of the therapeutic armamentarium.
Suggested Readings
- De Keersmaecker K, Cools J. Chronic myeloproliferative disorders: a tyrosine kinase tale. Leukemia 2006; 20:200–205.
- Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy in myeloproliferative disorders. Nat Rev Cancer 2007; 7:673–683.
- Tefferi A. The history of myeloproliferative disorders: before and after Dameshek [published online ahead of print September 20, 2007]. Leukemia 2008; 22:3–13. doi:10.1038/sj.leu.2404946.
- Tefferi A, Elliot M. Thrombosis in myeloproliferative disorders: prevalence, prognostic factors, and the role of leukocytes and JAK2V617F. Semin Thromb Hemost2007; 33:313–320.
- Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms [published online ahead of print September 20, 2007]. Leukemia 2008; 22:14–22. doi:10.1038/sj.leu.2404955.
References
- Talarico LD. Myeloproliferative disorders: a practical review. Patient Care 1998; 30:37–57.
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 Revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes [published online ahead of print April 8, 2009]. Blood 2009; 114:937–951.
- Baxter EJ, Scott LM, Campbell PJ, et al; Cancer Genome Project. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet2005; 365:1054–1061.
- Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007; 356:459–468.
- Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythermia [published online ahead of print March 22, 2007]. Blood 2007; 110:840–846.
- Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients [published online ahead of print July 25, 2006]. Blood 2006; 108:3472–3476.
- Bilgrami S, Greenbeg BR. Polycythemia rubra vera. Semin Oncol 1995; 22:307–326.
- Hoffman R, Benz EJ Jr, Shattil SJ, et al. Hematology: Basic Principles and Practice. 3rd ed. New York, NY: Churchill Livingstone; 2000:1106-1155, 1172-1205.
- Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel [published online ahead of print May 8, 2007]. Blood2007; 110:1092–1097.
- Tefferi A, Spivak JL. Polycythemia vera: scientific advances and current practice. Semin Hematol 2005; 42:206–220.
- Barbui T, Lunghi M, Tieghi A, et al. A large-scale trial testing the intensity of cytoreductive therapy to prevent cardiovascular events in patients with polycythemia vera (CYTO-PV trial). Abstract presented at: 54th ASH Annual Meeting and Exposition; December 8-11, 2012; Atlanta, GA. Abstract 4.
- Mesa RA, Verstovsek S, Cervantes F, et al; on behalf of the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT). Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): consensus on terminology by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT ) [published online ahead of print January 8, 2007]. Leuk Res 2007; 31:737–740.
- Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med 2000; 342:1255–1265.
- Hussein K, Pardanani A, Van Dyke DL, Hanson CA, Tefferi A. International Prognostic Scoring System-independent cytogenetic risk categorization in primary myelofibrosis [published online ahead of print November 9, 2009]. Blood 2010; 115:496–499.
- Chaligné R, Tonetti C, Besancenot R, et al. New mutations of MPL in primitive myelofibrosis: only the MPL W515 mutations promote a G1/ S-phase transition [published online ahead of print June 5, 2008]. Leukemia 2008; 22:1557–1566.
- Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment) [published online ahead of print December 14, 2009]. Blood 2010; 115:1703–1708.
- Tefferi A, Lasho TL, Jimma T, et al. One thousand patients with primary myelofibrosis: the Mayo Clinic experience. Mayo Clin Proc 2012; 87:25–33.
- Tefferi A. How I treat myelofibrosis [published online ahead of print January 3, 2011]. Blood 2011; 117:3494-3504.
- Begna KH, Pardanani A, Mesa R, et al. Long-term outcome of pomalidomide therapy in myelofibrosis [published online ahead of print November 12, 2011]. Am J Hematol 2012; 87:66–68.
- Barosi G, Hoffman R. Idiopathic myelofibrosis. Semin Hematol 2005; 42:248–258.
- Tefferi A, Cortes J, Verstovsek S, et al. Lenalidomide therapy in myelofibrosis with myeloid metaplasia [published online ahead of print April 11, 2006]. Blood2006; 108:1158–1164.
- Tabarroki A, Tiu RV. Immunomodulatory agents in myelofibrosis [published online ahead of print June 6, 2012]. Expert Opin Investig Drugs 2012; 21:1141–1154.
- Tefferi A, Mesa RA, Nagorney DM, Schroeder G, Silverstein MN. Splenectomy in myelofibrosis with myeloid metaplasia: a single-institution experience with 223 patients. Blood 2000; 95:2226–2233.
- Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012; 366:799–807.
- Tefferi A, Solberg LA, Silverstein MN. A clinical update on polycythemia vera and essential thrombocythemia. Am J Med 2000; 109:141–149.
- Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and high risk of thrombosis. N Engl J Med 1995; 332:1132–1136.
- Murphy S, Peterson P, Iland H, Laszio J. Experience of the Polycythemia Vera Study Group with essential thrombocythemia: a final report on diagnostic criteria, survival, and leukemic transition by treatment. Semin Hematol 1997; 34:29–39.
- Beer PA, Erber WN, Campbell PJ, Green AR. How I treat essential thrombocythemia [published online ahead of print November 24, 2010]. Blood 2011; 117:1472–1482.
- Cortelazzo S, Viero P, Finazzi G, D’Emilio A, Rodeghiero F, Barbui T. Incidence and risk factors for thrombotic complications in a historical cohort of 100 patients with essential thrombocythemia. J Clin Oncol 1990; 8:556–562.
- Harrison CN, Campbell PJ, Buck G, et al; for the United Kingdom Medical Research Council Primary Thrombocythermia 1 Study. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005; 353:33–45.