
Multiple Sclerosis
Carrie M. Hersh
Robert J. Fox
Published: June 2014
Multiple sclerosis (MS) is a chronic inflammatory disorder of the central nervous system (CNS) and is one of the most common causes of nontraumatic disability among young and middle-aged adults. MS-related healthcare costs are estimated to be more than $10 billion annually in the United States (US). As symptoms of MS are extremely variable and often quite subtle, diagnosis and management have been greatly enhanced by the development of magnetic resonance imaging (MRI). Therapies that slow progression of disease are available; therefore, early diagnosis and treatment are important in limiting the impact of this potentially devastating disease.
Definition
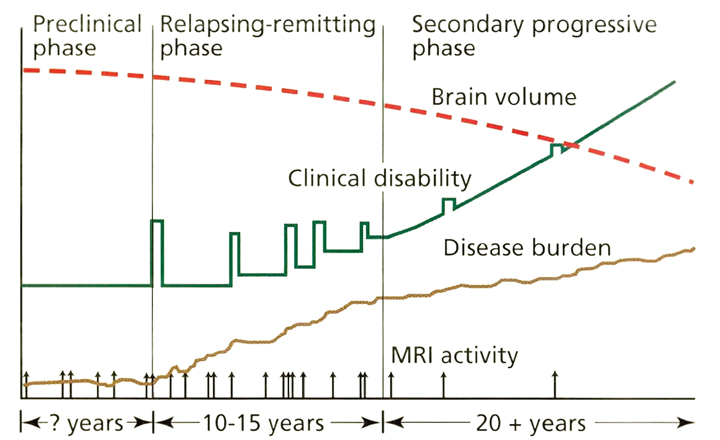
As outlined in Figure 1, there are several different forms of MS. Since these classifications were based upon clinical characteristics, they are empiric and do not reflect specific biologic pathophysiology. Nonetheless, they provide an organized framework for diagnosis and long-term management. Relapsing-remitting MS (RRMS) is the most common form of the disease, where symptoms appear for several days to weeks, after which they usually resolve spontaneously. After tissue damage accumulates over many years, patients often enter the secondary progressive stage of MS (SPMS), where pre-existing neurologic deficits gradually worsen over time. Relapses can be seen during the early stages of SPMS, but are uncommon as the disease further progresses. About 15% of patients have gradually worsening manifestations from the onset without clinical relapses, which defines primary progressive MS (PPMS). Patients with PPMS tend to be older, have fewer abnormalities on brain MRI, and generally respond less effectively to standard MS therapies.1 Progressive relapsing MS is defined as gradual neurologic worsening from the onset with subsequent superimposed relapses. Progressive relapsing MS (and possibly a proportion of PPMS) is suspected to represent a variant of SPMS, where the initial relapses were unrecognized, forgotten, or clinically silent.
Neuromyelitis optica (NMO), or Devic’s disease, is an uncommon demyelinating disorder, but common mimic of MS.2 NMO manifests as recurrent optic neuritis and longitudinally extensive transverse myelitis, typically extending over three or more vertebral segments. A relatively specific antibody–the NMO antibody–has been identified and recognizes the aquaporin-4 water channel in astrocyte foot processes located adjacent to capillary walls.3,4 NMO is a clinically aggressive disease with associated marked disability. In this context, recognition and diagnosis of NMO is important so that both acute and long-term therapies are appropriately initiated since they differ from treatments for MS.
Prevalence
MS affects approximately 400,000 people in the US and 2.5 million worldwide. In the US, prevalence estimates are approximately 90 per 100,000 population. MS symptoms can start anywhere between 10 and 80 years of age, but onset is usually between 20 and 40 years, with a mean of 32 years. Although MS is more frequently seen in Caucasians than African Americans, the latter group appears to accumulate disability more quickly, suggesting more destructive tissue injury in this population. The prevalence of MS varies by location and generally increases the further one travels from the equator in either hemisphere. It remains unclear whether this altered incidence represents an environmental influence, genetic difference, or variable surveillance.
Pathophysiology
Early in the disease course, MS involves recurrent bouts of CNS inflammation that results in damage to both the myelin sheath surrounding axons as well as the axons themselves. Histologic examination reveals foci of severe demyelination, decreased axonal and oligodendrocyte numbers, and gliotic scarring. The exact cause of inflammation remains unclear, but an autoimmune response directed against CNS antigens is suspected. Pathological studies suggest that different patients may have different etiologies for inflammation: some patients appear to have T-cell mediated or T-cell-plus-antibody-mediated autoimmune responses, while others have a primary disorder with the myelin-producing oligodendrocyte cells.5 This latter mechanism is reminiscent of virus- or toxin-induced demyelination rather than autoimmunity in this subset of patients. Further research is needed to understand how these different pathologic subtypes affect prognosis and response to treatments. Currently, brain biopsy is the only method to determine pathologic subtypes, but studies are underway to find blood, cerebrospinal fluid (CSF), and MRI markers.
Historically, MS was classified as an inflammatory disease targeting white matter, with diagnostics and therapeutics focused on this region of pathology. However, recent imaging and histopathological studies have suggested that cortical demyelination plays a crucial role in MS pathogenesis and cognitive dysfunction. Cortical demyelination is now recognized in early MS.6 Although some investigative MRI modalities capture some cortical involvement, including double inversion recovery sequences at 3 tesla (3T) and ultra-high field (7T) MRI, conventional MRI metrics used in clinical practice still do not show these changes. Likewise, extensive cortical demyelination that is seen in histopathological studies is not clearly picked up on any current MRI modality. This pathology/imaging discordance demonstrates that we are still technologically disadvantaged in accurately assessing cortical lesion pathology in the live patient.
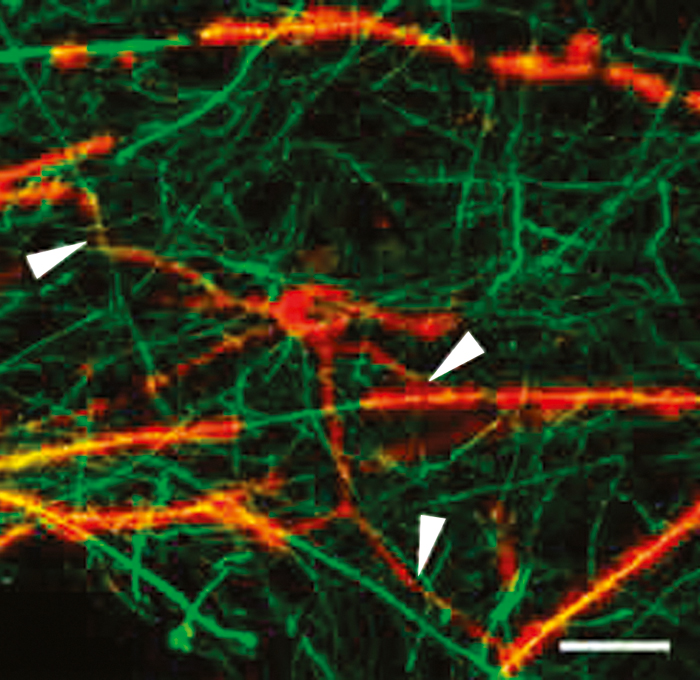
In the past, inflammation was thought to involve only demyelination, but pathologic studies have found significant axonal pathology, as well. In actively demyelinating MS lesions, an average of more than 11,000 transected axons/mm3 were observed, while control brain tissue had less than one transected axon/mm.7 Significant axonal injury is also observed in cortical demyelinating lesions. Clearly, axonal injury is significant in the early stages of disease.
Later in the disease course, gradual progression of disability is observed. However, there is significantly less active inflammation during this period, so clinical progression may arise instead from degenerative changes. Nonetheless, oligodendrocyte progenitor cells capable of remyelinating axons have been observed even in white matter plaques from patients with chronic MS (Figure 2).8 This observation suggests that the potential for remyelination persists even very late in the disease course, which is an encouraging indicator for possible therapeutic targets at this late stage of disease.
Current concepts of the pathophysiology of MS are illustrated in Figure 3. On average, patients have clinical relapses every 1 to 2 years during the relapsing-remitting phase of the disease. Serial MRI studies have shown that lesions develop up to 10 to 20 times more frequently than clinical relapses. Thus, although RRMS appears to have clinically active and quiescent periods, inflammatory lesions are developing and evolving almost continuously. A current hypothesis states that overt progression of disability (i.e., SPMS) occurs when ongoing irreversible tissue injury exceeds a critical threshold beyond which the nervous system can no longer compensate. It is thought that at this point the disease has become primarily a degenerative process with neurologic deterioration independent of ongoing inflammation, although superimposed inflammation can continue to cause additional injury.
An important implication of this hypothesis is that the accumulation of irreversible tissue damage limits the potential for benefit from disease-modifying immunomodulatory therapy as the disease progresses and becomes a degenerative process. To be maximally effective, disease-modifying therapy (DMT) should be started early in the relapsing-remitting phase and before permanent disability develops.
Signs and Symptoms
MS can cause almost any neurologic symptom since it can affect any area of the brain, optic nerve, and spinal cord. Disease localized to the spinal cord may cause sensory or motor changes involving one or both sides of the body or below a certain spinal cord level (i.e., hemiparesis or paraparesis). Brainstem involvement may present as diplopia, altered sensation or weakness of the face, or ataxia. Inflammation of the optic nerve (optic neuritis) usually presents as blurry vision with painful eye movements, and is often an early clinical manifestation of RRMS. Of all the lesions in MS, cerebral lesions are the most common but cause the least amount of symptoms. Most cerebral lesions are not located in eloquent regions, and so are clinically silent and identified only by brain MRI. Very large cerebral lesions may present with weakness or numbness, and rarely may cause aphasia or other cortical dysfunction. Symptoms of a clinical relapse typically arise over hours to days, worsen over several weeks, and then gradually subside over several weeks or months. Residual enduring neurologic symptoms are common. The gradual progression of progressive MS is most commonly seen as an evolving myelopathy causing asymmetric leg weakness, ataxia, and spasticity.
Other common symptoms of MS include bladder and bowel dysfunction, memory changes, fatigue, and affective disorders such as depression. Although these symptoms are not uncommon in MS, they are also very nonspecific and can be seen in a multitude of disorders.
Lhermitte’s sign is a nonspecific sign whereby flexion of the neck causes an electrical-like shooting sensation that extends into the arms or down the back. It is thought to arise from partially demyelinated tissue, whereby mechanical stimulation leads to axonal activation.
Diagnosis
There are no pathognomonic clinical, laboratory, or imaging findings in MS. The diagnosis ultimately is a clinical decision based upon weighing the factors that support the diagnosis against those that fail to support it or point to the possibility of an alternative diagnosis (i.e., NMO).
The Schumacher criteria from 1965 capture the essence of the diagnosis of MS: CNS lesions disseminated in space and time, and the elimination of alternative diagnoses.9 The Schumacher criteria required an age range between 10 and 50 years and objective abnormalities on examination, which are now outdated. However, the main concepts captured by these criteria remain relevant today.
The criteria from the International Panel on MS Diagnosis, also called the McDonald Criteria, are a set of diagnostic criteria for MS that incorporate the clinical characteristics alone or in combination with MRI features.10 Revisions were made in 2005 and again in 2010 as a reflection of an increased understanding of the natural history of MS revealed by the MRI and clinical progression, and improved MRI techniques. The latest version of the McDonald Criteria (2010) has simplified the diagnostic process, allowing MS to be diagnosed earlier (Table 1). After a single episode of demyelination, certain findings on one MRI can help fulfill the diagnostic criteria for MS, even before a second clinical episode or new MRI lesion. The revisions also preserve diagnostic sensitivity and specificity and address their applicability across different populations, allowing for more uniform and widespread use across groups.
Table 1: Summary of the McDonald Criteria (2010)*
| Clinical Findings at Presentation
|
||
|---|---|---|
| Episodes from History | Objective Clinical Signs | Additional Data Needed From MRI or Clinical Follow-up |
| 2 Attacks | 2 Lesions | None† |
| 2 Attacks | 1 Lesion | Evidence of DIS‡ |
| 1 Attack | 2 Lesions | Evidence of DIT§ |
| 1 Attack | 1 Lesion | Evidence of both DIS and DIT |
| Progressive course over 1 year | DIS demonstrated by two of:
|
|
CNS, central nervous system; CSF, cerebrospinal fluid; DIS, dissemination in space; DIT, dissemination in time; MRI, magnetic resonance imaging; MS, multiple sclerosis.
* Demonstration of DIS and DIT can be made by clinical features alone or by a combination of clinical and MRI features.
† Although no further data is needed to make the diagnosis of MS, MRI is typically obtained to both exclude other diagnoses and stage the severity of disease.
‡ DIS principle requires that there are asymptomatic lesions typical of MS present in two or more sites within the CNS: periventricular, subcortical, infrantentorial, and spinal cord.
§ DIT principle requires that two attacks separated by more than 30 days have occurred in different parts of the CNS. MRI criteria for DIT stipulate either an asymptomatic enhancing T2 lesion along with a non-enhancing T2 lesion on any one scan, or a new T2 or gadolinium-enhancing lesion on a follow-up scan.
In all cases, the practitioner must rule out better explanations for the clinical presentation other than multiple sclerosis.
Republished with permission of Demos Medical from Rae-Grant A, Fox RJ, Bethoux F. Multiple Sclerosis and Related Disorders: Clinical Guide to Diagnosis, Medical Management, and Rehabilitation. New York, NY: Demos Medical; 2013:51; permission conveyed through Copyright Clearance Center, Inc.
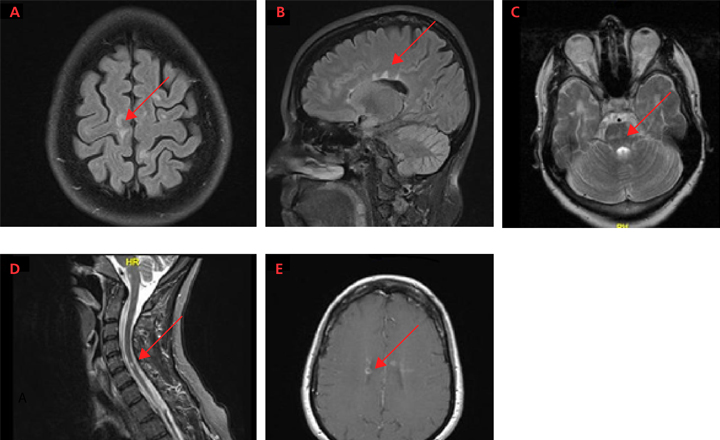
Although the diagnosis of MS cannot be based on an MRI alone, typical MRI lesions in the periventricular and subcortical regions, as well as the brainstem, cerebellum, and spinal cord can raise the suspicion of MS, warranting further diagnostic work-up or monitoring. MRI is typically obtained at the time of diagnosis to both exclude other diagnoses and stage the severity of disease. Patients with a typical history of MS without typical MRI findings are highly unusual and should prompt consideration of an alternative diagnosis.
Therapy
Management of MS requires multiple therapeutic approaches. The current goals of management of RRMS involve the treatment of acute relapses, prevention of new disease activity, and management of symptoms that affect the quality of life.
Acute Relapse
Several studies have found that treatment with corticosteroids can shorten the length of relapse and may even improve long-term outcome.11,12 A typical regimen is 500 to 1,000 mg of intravenous (IV) methylprednisolone followed by a tapering dose of oral prednisone over several weeks. The standard protocol at the Mellen Center, Cleveland Clinic, is IV methylprednisolone 1,000 mg daily for 3 days, followed by a 12-day prednisone taper (60 mg daily, decreasing every 4 days by 20 mg). Evaluation of a relapse should include a search for precipitating factors such as fever, upper respiratory illness, or bladder infection. For patients who do not respond sufficiently to corticosteroids or who do not tolerate corticosteroids, adrenocorticotropic hormone could be considered.
DMT for RRMS
After the acute relapse is treated, consideration should turn to DMT, which primarily targets the early inflammatory phase of disease. Current therapies target the immune dysfunction in MS and resultant neural tissue damage with the goal of preventing or at least reducing the long-term risk of clinically significant disease. Early treatment is key since it offers the greatest chance of preventing or delaying tissue injury and long-term disability. Although the underlying pathogenesis of MS still remains poorly understood, remarkable progress has been made in the development of drug therapies that inhibit disease activity. Using available options, including the advent of newer more effective drugs, there is the potential to achieve a disease-free status, characterized by the prevention of clinical relapses and disability progression and absence of new lesions on MRI. All of the DMTs are expensive and cost about $50,000 per year. Monitoring patients clinically and with surveillance MRI scans during treatment is important to detect nonresponders and modify therapy accordingly.
It is important to note that all current MS therapies are preventative and not restorative. As the disease progresses, response to DMT typically declines. The key to successful treatment of MS is to slow the inflammatory process early in the disease. It is likely that the accumulation of irreversible tissue damage limits the potential for benefit from DMT as the disease progresses. The therapeutic nihilism of the past should be replaced by aggressive treatment and monitoring, while carefully balancing the potential risks and benefits.
Injectable Platform Therapies
Five first-line injectable therapies, otherwise known as platform agents, are currently available in the US: intramuscular interferon (IFN) β-1a (Avonex, weekly injection), subcutaneous IFN β-1a (Rebif, thrice-weekly injection), subcutaneous IFN β-1b (Betaseron and Extavia, alternate-day injection), and glatiramer acetate (GA; Copaxone, daily vs thrice-weekly subcutaneous injection). The IFN medications are recombinant products with an amino acid sequence that is identical or nearly identical to that of human IFN β-1. GA is a random polypeptide based on the amino acid sequence of a myelin protein. All of these medications appear to modulate the immune response in MS, although GA and IFN medications probably work through different mechanisms.
In randomized, placebo-controlled trials, all of these medications were shown to decrease the rate of clinical relapses by about 30%, decrease the severity of the relapses, and have beneficial effects on measures of disease activity on MRI.13–16 All of the platform medications are generally well or moderately tolerated, and 15 to 20 years of accumulated data and clinical experience suggest strong long-term safety. The platform therapies are similar in efficacy, and selection is generally based on physician and patient preferences and side-effect profile. Potential adverse effects of the IFN medications include hepatic and hematological toxicities, flu-like side effects, and worsening of headaches, depression, and spasticity. GA may have the potential for more bothersome injection site reactions in thin patients. All four medications are appropriate first-line therapies in RRMS.
Oral Therapies
There are currently three oral DMTs approved by the U.S. Food and Drug Administration (FDA) to reduce disease activity in relapsing forms of MS. These therapies include fingolimod (Gilenya 0.5 mg, one tablet daily), teriflunomide (Aubagio 7 mg or 14 mg, one tablet daily), and dimethyl fumarate (Tecfidera 240 mg, one tablet twice daily).
Fingolimod. Fingolimod was approved in September 2010 as the first oral disease therapy for MS. Fingolimod acts by binding to the sphingosine-1-phosphate receptor on lymphocytes, which prevents egress of lymphocytes from lymph nodes. The sequestration of autoreactive lymphocytes prevents their recirculation to the CNS, thus inhibiting one of the primary steps in MS pathogenesis. Fingolimod may have direct effects within the CNS, as well. Fingolimod was shown to reduce relapse rate by 54% and disability progression by 30% in comparison with placebo, with MRI measures of disease activity supporting these clinical findings.17 In a head-to-head clinical trial against active comparator IFN beta-1a (Avonex), fingolimod demonstrated a greater reduction in annualized relapse rate (ARR, approximately 33%) and reduction in MRI disease activity, but no difference in disability progression.18
Despite its more potent clinical efficacy as compared with the platform medications, fingolimod is generally used as a second-line agent owing to its adverse-effect profile. During clinical trials, most of the reported side effects were mild to moderate in severity and included upper respiratory tract infections, headache, diarrhea, and back pain. The most concerning adverse effects included cardiac events (bradycardia and atrioventricular block at treatment initiation), elevated liver enzymes, fatal infections (herpes virus infections), and macular edema. The development of these serious side effects during clinical trials led to strict FDA recommendations for close monitoring during first dose administration and risk factor mitigation strategies to reduce potential serious complications. These parameters include baseline complete blood count (CBC), liver function tests (LFTs), electrocardiogram (ECG), ophthalmological evaluation, and serum varicella zoster virus (VZV) immunoglobulin G titer prior to fingolimod initiation. First-dose administration is conducted in a medical center where patients are monitored for 6 hours, with hourly vital sign checks and an ECG after 6 hours. Extended monitoring for a total of 24 hours is needed if bradycardia or QT prolongation exists, and with other cardiac risk factors. Periodic labs including CBC and LFTs and ophthalmological reassessment are used for continued safety surveillance.
Teriflunomide. Teriflunomide was the second oral DMT approved by the FDA and reached market approval in September 2012. It is an active metabolite of leflunomide and acts by inhibiting the de novo synthesis of pyrimidine nucleotides through the inhibition of dihydroorotate dehydrogenase,19 and inhibits T-lymphocyte activation and cytokine production in addition to cytostatic effects on proliferating B- and T-lymphocytes.20 Two phase III trials that compared teriflunomide against placebo showed modest reduction in ARR (TEMSO, 31% ARR reduction; TOWER, 36% ARR reduction).21,22 A phase III trial comparing teriflunomide and subcutaneous IFN beta-1b failed to show superiority.23
Teriflunomide was found to be relatively safe and generally well-tolerated in phase II and III clinical trials. There was no increase in opportunistic infections, and most of the adverse effects related to the medication were transitory. The most common adverse effects observed in the trials and in clinical practice are mild to moderate in severity and include gastrointestinal symptoms, decrease in hair density, mild elevations in liver enzymes, rash, and fatigue. A long-term safety study demonstrated that teriflunomide was safe over 8.5 years of follow-up.24 Leflunomide is highly teratogenic, and it is likely that teriflunomide carries similar fetal risks, and thus its pregnancy category X designation. In this context, pregnancy must be excluded in all women of childbearing potential, and effective contraceptive methods must be employed for both women and men. An accelerated removal process is available for patients who become pregnant or desire to become pregnant while taking teriflunomide. Risk mitigation strategies include LFTs at baseline and every 6 months while on the medication and a baseline and 6-month CBC. Testing for latent tuberculosis should also be conducted prior to drug initiation.
Dimethyl Fumarate. Dimethyl fumarate, otherwise known as BG-12, is the third oral DMT that was approved in April 2013. It is a fumaric acid ester with mechanism of action in MS that is thought to be related to its effect on nuclear factor-E2-related factor (Nrf) 2.25 Fumaric acid esters have been used in Europe to treat psoriasis under the trade name Fumaderm and are known to have a relatively favorable safety profile.26 Dimethyl fumarate demonstrated clinical efficacy in phase III clinical trials, with 48% to 53% ARR reduction as compared with placebo.27,28 Data also showed a reduction in MRI measures of activity,27 and reduction in risk of disability progression,27although the latter finding was not found to be significant in the CONFIRM trial.28
The most frequent adverse effects observed during trials with dimethyl fumarate were gastrointestinal symptoms, including stomach pain, nausea, vomiting, and diarrhea. Transient skin flushing can also be seen. Gastrointestinal symptoms are generally more prominent during the first several weeks of treatment and usually improve significantly thereafter. Concomitant use of aspirin substantially reduces associated flushing. There was no increased rate of serious adverse effects during phase III clinical trials, and infections were seen at a similar rate compared with placebo. Lymphopenia was seen in approximately 2% of patients on dimethyl fumarate in clinical trials, but there was no associated increased risk of infections. In this context, baseline and annual CBC are recommended surveillance measures while on this medication.
Natalizumab
Natalizumab, approved by the FDA in November 2004, is a monoclonal antibody (Ab) targeting the cellular adhesion molecule very late antigen-4 (VLA-4), administered by IV infusion every 4 weeks. By blocking VLA-4, fewer inflammatory cells enter the brain and thereby blunt CNS inflammation typical of MS. Results from phase III trials showed that natalizumab reduces clinical relapses by 67% and new brain lesions by 83%,29,30 making natalizumab the most clinically effective medicine for RRMS to date.
Natalizumab is relatively well-tolerated with mild headache, fatigue, anxiety, menstrual irregularities, peripheral edema, and routine infections (upper respiratory tract infection and pharyngitis) occasionally observed. Infusion-related hypersensitivity reactions (i.e., hives and pruritus) were observed in 2% to 4% of patients and are thought to represent immune-mediated hypersensitivity reactions. Anaphylactoid reactions were very rarely observed. Patients who demonstrate an infusion reaction should discontinue natalizumab immediately and not be re-treated.
The most concerning serious adverse effect of natalizumab is progressive multifocal leukoencephalopathy (PML), which occurs at an overall incidence of 2.1 per 1,000 population.31–33 PML is a serious viral infection of the brain, arising from the ubiquitous John Cunningham virus (JCV), which resides in the kidneys and bone marrow in about half of adults. Three identified risk factors that substantially alter an individual’s risk of PML include duration of natalizumab treatment, prior history of immunosuppressive therapy, and serum anti-JCV Ab status. In patients with natalizumab treatment exceeding 24 months, prior use of immunosuppressant drugs, and positive serum anti-JCV Ab testing, the risk for PML is estimated to be as high as 1:48. In this context, careful risk stratification prior to treatment initiation is recommended, which enables more informed clinical decision-making with regard to natalizumab use. It remains unknown whether PML can be effectively treated in natalizumab-related MS patients, though attempts to remove natalizumab from the blood (i.e., through plasmapheresis or leukopheresis) likely accelerates immune reconstitution and is recommended in patients with natalizumab-related PML. Because of PML, natalizumab was withdrawn from clinical use in February 2005 but received a second FDA approval in June 2006. Due to serious potential for PML, natalizumab is not generally considered a first-line therapy for MS and is generally reserved for patients who respond suboptimally or do not tolerate the MS therapies described above. However, the growing experience of JCV serology testing suggests that in subjects with persistently negative JCV serology, natalizumab could be considered as a first-line therapy. Discussions with potential natalizumab recipients should review the risks and potential benefits of this medication.
Changing DMT
Broadly speaking, it is appropriate to consider switching DMT for two reasons: breakthrough disease and intolerable adverse effects. There is no formal consensus on the optimal approach to either defining or managing breakthrough disease, although it is commonly encountered in the clinical setting. Breakthrough disease is generally defined as continued clinical or radiographic evidence of MS-related inflammatory disease activity despite being on an established DMT. Continued clinical relapses or new MRI lesions, particularly after 6 months of treatment with an established DMT, often constitutes breakthrough disease activity. The severity of relapses, their subsequent recovery, and the number and size of new or active MRI lesions all contribute to defining when patients are considered to have breakthrough disease activity. Continued surveillance of clinical and radiographic measures of disease activity is crucial during treatment, and it is generally recommended that patients are followed every 3 to 12 months with repeat brain MRI every 6 to 12 months, depending on the patient’s baseline disease status and DMT of choice.
Mitoxantrone (Novantrone) is a chemotherapy medication with demonstrated efficacy in very active relapsing and progressive MS.34 Administration is via IV infusion every 3 months, although a monthly induction course is sometimes used in patients with very active disease. Infusion side effects include nausea and alopecia. However, owing to the potential for long-term toxicities including cardiac injury that is cumulative over time and lymphoproliferative disorders including leukemia, it is now rarely used in treating MS. Cardiac injury can occur years after completing therapy, which warrants surveillance echocardiogram or multigated acquisition scans prior to each infusion and after the medication has been discontinued.
With respect to adverse effects, every attempt should be made to manage them symptomatically. However, if the patient remains intolerant of these adverse effects, change to another DMT should be considered. Table 2 summarizes common adverse effects and risk mitigation strategies for various DMTs.
Table 2: Summary of Current FDA-Approved DMTs for RRMS
| Generic Drug (Trade Name) | Dosing | Adverse Effects | Lab Monitoring and Risk Mitigation Strategies |
|---|---|---|---|
| Glatiramer acetate (Copaxone) | 20 mg SC daily |
|
None required |
| Interferon β-1a |
|
|
|
| (Avonex) | 30 µg IM weekly | ||
| (Rebif) | 44 µg SC 3 times weekly | ||
| Interferon β-1b |
|
|
|
| (Betaseron) | 0.25 mg SC qod | ||
| (Extavia) | 0.25 mg SC qod | ||
| Natalizumab (Tysabri) | 300 mg IV monthly |
|
Prior to initiation:
In all patients:
|
| Fingolimod (Gilenya) | 0.5 mg PO daily |
|
|
| Teriflunomide (Aubagio) | 7 mg or 14 mg PO daily |
|
|
| Dimethyl fumarate (Tecfidera) | 240 mg PO bid |
|
|
Ab, antibody; bid, twice daily; BP, blood pressure; CBC, complete blood count; CSF, cerebrospinal fluid; DMTs, disease-modifying therapies; ECG, electrocardiogram; FDA, U.S. Food and Drug Administration; IM, intramuscular; IV, intravenous; JCV, John Cunningham virus; LFTs, liver function tests; mg, milligram; MRI, magnetic resonance imaging; µg, microgram; PCR, polymerase chain reaction; PML, progressive multifocal leukoencephalopathy; PO, oral; qod, every other day; RRMS, relapsing-remitting multiple sclerosis; SC, subcutaneous; VZV, varicella zoster virus.
Republished with permission of Demos Medical from Rae-Grant A, Fox RJ, Bethoux F. Multiple Sclerosis and Related Disorders: Clinical Guide to Diagnosis, Medical Management, and Rehabilitation. New York, NY: Demos Medical; 2013:104; permission conveyed through Copyright Clearance Center, Inc.
Treatment for Progressive MS
Treatment of SPMS is more difficult than relapsing MS. Although DMTs were found in some trials to prevent progression of disability in SPMS, the effect was modest and seen primarily in those subjects with superimposed active inflammation. It appears worthwhile to use these anti-inflammatory medications during this stage if there is evidence of persistent active inflammation (either clinically or radiologically) and side effects are tolerated, but caution regarding reasonable clinical expectations is appropriate.
There are currently no treatments with demonstrated clinical efficacy in SPMS or PPMS in the absence of active inflammation. Two clinical trials provided evidence that intermittent pulses of IV methylprednisolone can help slow the progression of clinical disability in some patients.11,12 In light of the paucity of effective treatments, several national and international phase II and III clinical trials investigating potential therapies for progressive forms of MS are currently underway.
Role for Immunosuppressant Drugs in MS
Cyclophosphamide, methotrexate, azathioprine, and cyclosporine all have been studied in small- to medium-sized trials. The Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and MS Council for Clinical Practice Guidelines have made recommendations regarding these therapies.35Methotrexate, azathioprine, and cyclosporine were each found to be possibly effective (type C recommendation) in altering the course of disease, but cyclosporine was found to have an unacceptable risk-benefit ratio. In their review, pulse cyclophosphamide treatment did not alter the course of MS (type B recommendation), although a more recent clinical trial showed reduced relapses and MRI lesions in patients treated with cyclophosphamide.36 Given the 10 FDA-approved therapies for relapsing MS and relatively weak evidence supporting the efficacy of immunosuppressant therapies, they are not used frequently in the treatment of MS.
Symptomatic Therapies
Besides neurologic disability, MS can produce a variety of other symptoms that can interfere with daily activities. Identification and treatment of these symptoms should be considered throughout the disease course (Table 3). Specific recommendations for management of fatigue and urinary dysfunction have been outlined by the Multiple Sclerosis Council for Clinical Practice Guidelines. Aggressive evaluation and treatment for these and other symptoms of MS can improve quality of life significantly and are an important component of long-term management of patients with MS.
Table 3: Common Symptoms in MS and Potential Treatments
| Symptom | Pharmacotherapy | Miscellaneous |
|---|---|---|
| Fatigue | Amantadine 100 mg bid Pemoline 75 mg daily for two 4-week periods Modafinil 200-400 mg/day Armodafinil 150-250 mg/day |
|
| Depression | SSRIs, SNRIs, bupropion, mirtazapine, TCAs | Psychotherapy
Associated factors
|
| Bladder dysfunction | Anticholinergic medications for treatment of overactive bladder symptoms: Oxybutynin 5, 10, 15 mg up to tid Tolterodine 2, 4 mg bidAlpha-adrenergic blockers for treatment of impaired bladder emptying: Tamsulosin 0.4 mg qhs Doxazosin 1, 2, 4, 8 mg qhs Terazosin 1, 2, 5, 10 mg qhs |
Important factors to assess include:
Additional/nonpharmaceutical treatments:
|
| Bowel dysfunction | Bulk-forming agents: methylcellulose, psyllium hydrophilic mucilloid, guar gum Stool softeners: docusate Osmotic laxatives: lactulose, polyethylene glycol solution |
|
| Spasticity | Baclofen 5-10 mg/day, max 80 mg/day in 3-4 div doses Tizanidine 2-4 mg/day, max 36 mg/day in 3-4 div doses Diazepam 2 mg qhs, max 30 mg/day in 3-4 div doses Clonazepam 0.5 mg qhs, max 2 mg/day Gabapentin 100-300 mg/day, max 3,600 mg/day in 3-4 div doses |
|
| Sexual dysfunction | Sildenafil and other agents for men† |
|
bid, twice daily; div, divided; DMT, disease modifying therapy; g, grams; max, maximum; mg, milligrams; MRI, magnetic resonance imaging; MS, multiple sclerosis; qhs, at bedtime; SNRIs, serotonin norepinephrine reuptake inhibitors; SSRI, selective serotonin reuptake inhibitors; TCAs, tricyclic antidepressants; tid, three times daily; UTIs, urinary tract infections.
* The sacral neurostimulation device precludes the ability to undergo MRI studies, limiting its use in patients with MS where monitoring MRIs are often an integral part of patient management.
† Evidence for efficacy for sexual dysfunction in women with MS has been negative.
Outcomes
MS is a heterogeneous disease with a variable clinical course. Patients can progress rapidly over several years to death or may have a few relapses and then remain clinically stable for many decades. The accumulation of disability in MS is slower than previously thought and varies widely between individuals. Early studies reported a relatively quick progression from disease onset to walking with a cane, with a median time of about 15 years. However, more recent natural history studies have reported a longer time to reaching this disability milestone, with a median time from onset to cane of about 30 years. Likewise, in PPMS, early studies reported short median time from disease onset to cane of less than 10 years, whereas more current studies show that median time is closer to 15 years.37 The advent of effective immunomodulating therapy for relapsing MS may in part explain the transition to MS having a better long-term prognosis.
It is difficult to predict which patients will progress and which patients will remain relatively stable over time. Although there are clearly patients in whom the disease remains benign, it is very difficult to predict which patients will eventually follow this course. There are several prognostic factors for unfavorable clinical outcomes. Older age at onset, initial symptoms involving cerebellar, spinal cord or pyramidal systems, and higher initial clinical activity (i.e., high attack frequency and increased disability progression in the first 5 years) are all unfavorable prognostic factors. Prognostic radiologic measures include brain and spinal cord atrophy and gadolinium-enhancing lesions. MRI measures are also useful tools when evaluating the effect of MS therapies.38 Smoking and low serum vitamin D levels have emerged recently as additional predictors of poor long-term outcome.
Pregnancy does not seem to have any detrimental effects on the overall disease course of MS. Efficient family planning with the help of the obstetrician can help minimize the amount of time off of DMT. Pregnancy during MS is associated with a decreased incidence of relapses, but there is a rebound in relapse frequency postpartum.39 Relapses during pregnancy can be treated with short courses of high-dose corticosteroids if needed, though it is preferable to clinically monitor mild relapses since some adverse effects to glucocorticoids have been described. A mid-pregnancy visit with the treating neurologist is recommended for postpartum planning. It is also generally recommended that patients who were previously treated with DMT prior to pregnancy resume treatment immediately postpartum unless they plan to breastfeed. If breastfeeding is pursued, cranial MRI 2 months after delivery for disease surveillance is appropriate. If there is evidence of active disease, the patient should be counseled on resuming DMT.
Unfortunately, no DMT is proven to be safe during pregnancy or while breastfeeding, and is generally not recommended. Most of the available DMTs, including IFN, fingolimod, dimethyl fumarate, and natalizumab are pregnancy category C. Ongoing use of IFNs in women planning pregnancy may also be relatively contraindicated due to possible abortifacient properties. GA, pregnancy category B, appears to be the only available treatment that can be used with relative safety in women who need to continue DMT.40 As stated above, teriflunomide is pregnancy category X and should not be used in women of childbearing potential without effective contraception and counseling.
The effect of vaccines on MS has been studied very carefully, and there appears to be no adverse effect of vaccines on the course of disease.41 Vaccines can be given safely in MS and should be administered when clinically indicated. Of note, killed vaccine preparations are preferred over live preparations.
Summary
- Multiple sclerosis is a chronic inflammatory disorder affecting the brain, optic nerve, and spinal cord.
- Symptoms of multiple sclerosis can involve almost any neurologic function, so accurate diagnosis includes complete history, neurological examination, magnetic resonance imaging, and sometimes cerebrospinal fluid analysis.
- Many therapies are available to decrease the clinical episodes of inflammation, slow progression of disability, and ameliorate the symptoms from previous injury. Ongoing studies are currently investigating agents for progressive forms of multiple sclerosis.
- Early diagnosis and treatment are of key importance since effective treatment is difficult after the patient progresses into the later stages of multiple sclerosis.
References
- Cottrell DA, Kremenchutzky M, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study. 5. The clinical features and natural history of primary progressive multiple sclerosis. Brain 1999; 122:625–639.
- Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006; 66:1485–1489.
- Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364:2106–2112.
- Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel [published online ahead of print August 8, 2005]. J Exp Med 2005; 202:473–477. doi:10.1084/jem.20050304.
- Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassman H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000; 47:707–717.
- Lucchinetti CF, Popescu BF, Bunyan RF, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med 2011; 365:2188–2197.
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998; 338:278–285.
- Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med 2002; 346:165–173.
- Schumacker GA, Beebe G, Kibler RF, et al. Problems of experimental trials of therapy in multiple sclerosis: report by the panel on the evaluation of experimental trials of therapy in multiple sclerosis. Ann N Y Acad Sci 1965; 122:552–568.
- McDonald WI, Compston DA, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol 2001; 50:121–127.
- Milligan NM, Newcombe R, Compston DA. A double-blind controlled trial of high dose methylprednisolone in patients with multiple sclerosis: 1. clinical effects. J Neurol Neurosurg Psychiatry 1987; 50:511–516.
- Sellebjerg F, Frederiksen JL, Nielsen PM, Olesen J. Double-blind, randomized, placebo-controlled study of oral, high-dose methylprednisolone in attacks of MS. Neurology 1998; 51:529–534.
- Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol 1996; 39:285–294.
- Johnson KP, Brooks BR, Cohen JA, et al; Copolymer 1 Multiple Sclerosis Study Group. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. Neurology 1995; 45:1268–1276.
- PRISMS Study Group. Randomized double-blind placebo-controlled study of interferon β-1a in relapsing/remitting multiple sclerosis. Lancet 1998; 352:1498–1504.
- IFNB Multiple Sclerosis Study Group, University of British Columbia MS/MRI Analysis Group. Interferon beta-1b in the treatment of multiple sclerosis: final outcome of the randomized controlled trial. Neurology 1995; 45:1277–1285.
- Kappos L, Radue EW, O’Connor P, et al; FREEDOMS Study Group. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis [published online ahead of print January 20, 2010]. N Engl J Med 2010; 362:387–401. doi:10.1056/NEJMoa0909494.
- Cohen JA, Barkhof F, Comi G, et al; TRANSFORMS Study Group. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis [published online ahead of print January 20, 2010]. N Engl J Med 2010; 362:402–415. doi:10.1056/NEJMoa0907839.
- Greene S, Watanabe K, Braatz-Trulson J, Lou L. Inhibition of dihydroorotate dehydrogenase by the immunosuppressive agent leflunomide. Biochem Pharmacol1995; 50:861–867.
- Xu X, Blinder L, Shen J, et al. In vivo mechanism by which leflunomide controls lymphoproliferative and autoimmune disease in MRL/MpJ-lpr/lpr mice. J Immunol1997; 159:167–174.
- O’Connor P, Wolinsky JS, Confavreux C, et al; TEMSO Trial Group. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med 2011; 365:1293–1303.
- Genzyme reports positive top-line results of TOWER, a pivotal phase III trial for AubagioTM (teriflunomide) in relapsing multiple sclerosis. Genzyme website. news.genzyme.com/press-release/…. Published June 1, 2012. Accessed May 29, 2014.
- Vermersch P, Czlonkowska A, Grimaldi LM, et al; TENERE Trial Group. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial [published online ahead of print October 14, 2013]. Mult Scler 2014; 20:705–716. doi:10.1177/1352458513507821.
- Confavreux C, Li DK, Freedman MS, et al; Teriflunomide Multiple Sclerosis Trial Group. Long-term follow-up of a phase 2 study of oral teriflunomide in relapsing multiple sclerosis: safety and efficacy results up to 8.5 years [published online ahead of print February 3, 2012]. Mult Scler 2012; 18:1278–1289. doi:10.1177/1352458512436594.
- Linker RA, Lee D-H, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain2011; 134:678–692.
- Reich K, Thaci D, Mrowietz U, Kamps A, Neureither M, Luger T. Efficacy and safety of fumaric acid esters in the long-term treatment of psoriasis—a retrospective study (FUTURE). J Dtsch Dermatol Ges 2009; 7:603–611.
- Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med 2012; 367:1098–1107.
- Fox R, Miller D, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med 2012; 367:1087–1097.
- Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med 2006; 354:911–923.
- Polman CH, O’Connor PW, Hardova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006; 354:899–910.
- Sorensen PS, Bertolotto A, Edan G, et al. Risk stratification for progressive multifocal leukoencephalopathy in patients treated with natalizumab. Mult Scler 2012; 18:143–152.
- Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med 2012; 366:1870–1880.
- Yousry TA, Major EO, Ryschkewitsch C, et al. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. N Engl J Med2006; 354:924–933.
- Hartung HP, Gonsette R, the MIMS Study Group. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, randomised, multicentre trial. Lancet 2002; 360:2018–2025.
- Goodin DS, Frohman EM, Garmany GP, et al. Disease modifying therapies in multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Neurology 2002; 58:169–178.
- Smith DR, Weinstock-Guttman B, Cohen JA, et al. A randomized blinded trial of combination therapy with cyclophosphamide in patients with active multiple sclerosis on interferon beta. Mult Scler 2005; 11:573–582.
- Tremlett H, Zhao Y, Rieckmann P, Hutchinson M. New perspectives in the natural history of multiple sclerosis. Neurology 2010; 74:2004–2015.
- Rudick RA, Lee J-C, Simon J, Ransohoff RM, Fisher E. Defining interferon b response status in multiple sclerosis patients. Ann Neurol 2004; 56:548–555.
- Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T, the Pregnancy in Multiple Sclerosis Group. Rate of pregnancy-related relapse in multiple sclerosis. N Engl J Med 1998; 339:285–291.
- Cree BAC. Update on reproductive safety of current and emerging disease-modifying therapies for multiple sclerosis. Mult Scler 2013; 19:835–843.
- Confavreux C, Suissa S, Saddier P, Bourdes V, Vukusic S, the Vaccines in Multiple Sclerosis Study Group. Vaccinations and the risk of relapse in multiple sclerosis. N Engl J Med 2001; 344:319–326.